





1. Background
The most distinctive disorders within the pediatric disease spectrum are caused by aberrant developmental processes, including numerous difficult-to-diagnose hereditary respiratory diseases that are predominantly monogenic disorders. Fortunately, due to increasing awareness of disease-related gene variants and improved molecular diagnostic techniques, many rare respiratory diseases with complex clinical phenotypes can now be diagnosed (1). Proband-only medical exome sequencing has been reported to correctly diagnose up to 28% of suspected genetic cases in China (2). In a previous study (3), the sequenced genes of 97 children with difficult-to-diagnose respiratory diseases yielded an overall diagnostic rate of 32.0%. The diagnostic process usually begins with symptom assessments conducted by pulmonary disease specialists, who generally base their diagnoses on commonly observed symptoms of prevalent respiratory diseases (i.e., asthma, pneumonia, bronchitis, and bronchiectasis). Diagnosing pediatric intractable respiratory diseases is challenging due to the diverse clinical and genetic factors. Genetic testing allows pediatric clinicians to accurately diagnose and begin treatment earlier, particularly in resource-limited medical institutions lacking other diagnostic strategies. A study at Beijing Children's Hospital (4) demonstrated that exome sequencing is an effective first-tier test for diagnosing monogenic respiratory diseases in China.
2. Objectives
The current retrospective study was performed on 24 children with difficult-to-diagnose respiratory diseases who failed to respond to conventional treatment. The results are expected to enhance awareness regarding the phenotype and significance of molecular diagnostic methods, thus promoting their clinical application to improve the lives of children with monogenic respiratory diseases.
3. Methods
3.1. Patients
Twenty-four children admitted to the respiratory department of our hospital with clinically difficult-to-diagnose respiratory diseases from January 2018 to October 2022 were enrolled in the study. The inclusion criteria were as follows: Children with difficult-to-diagnose hereditary respiratory diseases; onset of respiratory disease manifestations prior to 16 years of age; family members or children provided consent to receive genetic testing. Compound heterozygosity was confirmed through parental testing if both parents were available. If not, it was inferred using population genetics, clinical phenotype, and bioinformatics data. The study was approved by the Institutional Review Board of our hospital [number (2020)-007].
3.2. Clinical Data Collection
For each child enrolled in the study, clinical data were collected, including general information, age of disease onset, course of disease, birth history, medical history, auxiliary examination data, genetic testing results, etc. Bronchial mucosal biopsy was carried out at least 8 weeks after acute infection.
3.2.1. Genetic Testing
Genomic DNA extraction from peripheral blood was conducted using a blood genome column medium extraction kit (Kangweishiji, China), according to the manufacturer’s instructions. Liquid hybridization of genomic DNA was performed using a Roche Nimble GenSeq Cap EZ Exome Library Kit v2.0 and SeqCap EZ Exomev2.0 Exome Enrichment Kit to capture probes (Roche, USA). For each sample, target DNA fragments were enriched to construct an exome library covering 19,119 genes, including whole exons and partial introns. High-throughput sequencing was performed using an Illumina NovaSeq 6000 series sequencer (PE150) to obtain sequences of ≥ 99% similarity to the target sequences.
3.2.2. Bioinformatics Analysis
Nonsynonymous substitutions and single-nucleotide polymorphisms (SNPs) with minor allele frequency (MAF) values of < 5% were filtered out using SIFT. Thereafter, the functional and pathogenic characteristics of mutated genes were analyzed by comparing the gene sequences to reference disease sequences within dbSNP, 1000 Genomes Project, ExAC, ESP, OMIM, Swiss Var, HGMD, ClinVar, and other databases. Single-base genetic variants with unknown pathogenicity were screened using Provean, SIFT, Polyphen2-HVAR, Polyphen2-HDIV, Mutation Taster, and other protein structure-based prediction software tools. In addition, MaxEntScan was used to screen sequences for potential alternate splice sites. Sanger sequencing confirmation was performed for all pathogenic, likely pathogenic, and variant of uncertain significance (VUS) reported in this study. Variants classified as pathogenic or likely pathogenic according to the ACMG guidelines were identified (5).
3.3. Statistical Analysis
Quantitative data are presented as the mean ± SD for continuous values, percentage values (%) for categorical data, and median and extreme values for non-parametric data.
4. Results
4.1. Demographic Data and Clinical Manifestations
A total of 24 children (14 males, 10 females) were recruited for this study. Due to the small sample size and outcome distribution, comparative and logistic regression analyses were avoided. Instead, the patients’ clinical features were described to explore potential associations. Overall, the mean age of symptom onset was 3.81 ± 3.16 years (range: 0 to 11 years) and the mean age at diagnosis was 2.32 ± 2.28 years (range: 1 month to 10 years). Twenty-two cases (91.67%) exhibited cough and recurrent expectoration of yellowish-brown or green purulent sputa; 10 cases (41.67%) exhibited recurrent wheezing; and 7 cases exhibited signs of malnutrition (29.17%), including 3 cases with severe malnutrition and 4 cases with moderate malnutrition. Other clinical features included electrolyte disorders, rhinitis, sinusitis, otitis media, hearing loss, total visceral inversion (Figure 1A), gastroesophageal reflux, allergic purpura, ichthyosis, premature delivery, anemia, tracheal diverticulum, and severe pneumonia (Table 1).
 images: A, atelectasis of left upper lobe, right heart, and total visceral inversion (case 4); B, abundant viscous sputum as seen under bronchoscope (case 1); C, abundant thick sputum observed under bronchoscope (case 8); D, bronchial mucosal biopsy (case 8); E, chest CT image showing extensive bronchiectasis (case 3); F, CT reconstruction showing bronchopulmonary dysplasia-induced airway changes (case 16)")
Representative bronchoscopy and computed tomography (CT) images: A, atelectasis of left upper lobe, right heart, and total visceral inversion (case 4); B, abundant viscous sputum as seen under bronchoscope (case 1); C, abundant thick sputum observed under bronchoscope (case 8); D, bronchial mucosal biopsy (case 8); E, chest CT image showing extensive bronchiectasis (case 3); F, CT reconstruction showing bronchopulmonary dysplasia-induced airway changes (case 16)
| NO | Sex | Age of Onset of Symptoms (y) | Disease Courses (y) | Respiratory Symptoms | Bronchiectasis | Nutritional Status | Bacterial Colonies | Other Clinical Manifestations |
|---|---|---|---|---|---|---|---|---|
| 1 | F | 2 | 4 | Recurrent cough, expectoration | Yes | Moderate malnutrition | Pa, SA | - |
| 2 | M | 0.5 | 0.83 | Recurrent cough, expectoration | None | Severe malnutrition | Normal | Electrolyte disturbance |
| 3 | F | 3 | 6 | Recurrent cough, expectoration, wheezing | Yes | Normal | Pa, Hi | - |
| 4 | M | 0 | 0.25 | Recurrent cough, expectoration | Yes | Normal | SP | Total visceral inversion, sinusitis, hearing abnormality, ventricular septal defect, pulmonary hypertension, intestinal malrotation |
| 5 | F | 0.5 | 3 | Recurrent cough, expectoration | None | Moderate malnutrition | Normal | Sinusitis, gastroesophageal reflux, gastritis |
| 6 | F | 5 | 1 | Recurrent cough, expectoration, wheezing | None | Normal | Normal | Rhinitis, suppurative otitis media |
| 7 | F | 0.42 | 5 | Recurrent cough, expectoration, wheezing | None | Normal | Normal | Sinusitis |
| 8 | M | 4 | 10 | Recurrent cough, expectoration | None | Normal | Normal | Sinusitis, otitis media, decreased serum immunoglobulin |
| 9 | F | 6 | 1 | Cough, expectoration | Yes | Severe malnutrition | Normal | Poor growth development, multiple fever and convulsions, IgA < 0.07g/L |
| 10 | M | 0.42 | 0.17 | Recurrent cough, expectoration, wheezing | None | Normal | SP | - |
| 11 | M | 0.5 | 2 | Recurrent cough, expectoration, wheezing | Yes | Normal | Normal | Tracheal diverticulum |
| 12 | F | 7 | 3 | Recurrent cough, expectoration | Yes | Normal | Normal | Allergic purpura |
| 13 | F | 6 | 1.5 | Recurrent cough, expectoration | Yes | Normal | Normal | Rhinitis |
| 14 | M | 7 | 4 | Recurrent cough, expectoration | Yes | Normal | Hi | Adenoid hypertrophy |
| 15 | M | 0.67 | 0.42 | Recurrent cough, expectoration | None | Normal | Normal | Premature birth, bronchopulmonary dysplasia |
| 16 | M | 0.67 | 0.42 | Recurrent cough, expectoration | None | Normal | Normal | Premature birth, bronchopulmonary dysplasia |
| 17 | M | 8 | 0.17 | Recurrent cough, expectoration | Yes | Moderate malnutrition | Normal | Rhiniti, moderate anaemia, bronchial stenosis |
| 18 | M | 11 | 3 | Recurrent cough, wheezing | Yes | Normal | Normal | Suboptimal asthma control |
| 19 | M | 4 | 2 | Recurrent cough, expectoration | None | Normal | Normal | - |
| 20 | F | 5 | 1 | Recurrent cough, expectoration, wheezing | Yes | Moderate malnutrition | Normal | Severe pneumonia requiring mechanical ventilation |
| 21 | M | 2.42 | 4 | Recurrent cough, expectoration, wheezing | Yes | Normal | Normal | Onset-severe |
| 22 | M | 10 | 0.17 | Recurrent cough, expectoration, hemoptysis | Yes | Severe malnutrition | Normal | Rhinitis , ichthyosis |
| 23 | F | 1.25 | 1 | Recurrent cough, expectoration, wheezing | None | Normal | Normal | None |
| 24 | M | 2.5 | 1.83 | Recurrent cough, expectoration, wheezing | None | Normal | Normal | Premature birth, bronchopulmonary dysplasia, rhinitis |
. Clinical Characteristics of 24 Chinese Pediatric Patients with Challenging-to-Diagnose Respiratory Diseases
4.2. Bronchoscopy and Imaging Examination Findings
Nineteen children (79.17%) underwent bronchoscopy, revealing tracheobronchial intimal inflammation and obstructed airways with abundant secretions (Figure 1B and C). Subsequently, 5 patients underwent bronchial mucosal biopsy (Figure 1D), which revealed an absence of inner and outer dynein arms in 1 patient (case 3). All 24 patients underwent high-resolution computed tomography (HRCT), which revealed bronchiectasis in 14 patients (58.33%; Figure 1E).
4.3. Laboratory Testing Results
Twenty-three cases (95.83%) submitted sputa or bronchial lavage fluid (BALF) specimens for culture, with positive bacterial cultures obtained for 5 cases (21.74%), including 1 cystic fibrosis (CF) case, 2 primary ciliary dyskinesia (PCD) cases, and 1 X-linked severe combined immunodeficiency (X-SCID) case. Test results obtained for these 5 cases revealed positive Pseudomonas aeruginosa culture results for 2 cases, positive Streptococcus pneumoniae results for 2 cases, positive Haemophilus influenzae results for 2 cases, and a positive Staphylococcus aureus result for 1 case. Further, 12 children (50%) completed pulmonary function testing. This testing was only performed for 12 children due to age limitations, as reliable testing requires patient cooperation, and as such, this test is generally difficult to perform in children under 5 - 6 years of age. Ten children (83.33%) exhibited signs of obstructive ventilatory dysfunction. Among them, 9 children (75%) exhibited mild symptoms, and 1 child (8.33%) exhibited moderately severe symptoms, while 3 children (25%) exhibited signs of mixed ventilatory dysfunction and 4 children (33.33%) had positive bronchodilation test results.
4.4. Genetic Testing Results
Traditional diagnostic methods were inconclusive; thus, genetic testing was chosen due to the presence of persistent symptoms. The genetic test results revealed abnormal gene sequences in 11 cases (Table 2). Of these, 2 were CF cases, 4 were PCD cases, and 5 were immunodeficiency disease cases, including 1 X-linked agammaglobulinemia (XLA) case, 1 ataxia-telangiectasia (A-T) case, 1 X-SCID case, 1 immunodeficiency-80 case, and 1 case with common variable immunodeficiency-8 with autoimmunity (Table 3). According to the reported characteristics of carriers of CFTR variants, diagnoses obtained here for 2 cases were consistent with a CF diagnosis, based on the clinical phenotypes and genetic testing results showing genotypes of the c.263T>G /c.1766 + 5G > T mutation (No.1) and homozygous c.1766 + 5G > T mutation (No.2) in CFTR genes. Meanwhile, 2 cases (identical twins) were found to carry c.1304T > C and c. 4098C > G CTFR mutations that they inherited from their phenotypically normal mother. They were ultimately diagnosed with bronchopulmonary dysplasia (Figure 1F). In addition, 1 child harboring a CTFR c.3205G > A mutation was diagnosed with asthma. Of the 13 genetically negative patients, 3 were clinically diagnosed with asthma, 3 with bronchiectasis post-infection, and 2 remain of unknown etiology. These 2 are under ongoing evaluation with planned functional studies, including sweat chloride testing when available. Further, 3 were diagnosed with BPD, 1 was diagnosed with PCD through bronchial mucosal biopsy, and 1 was diagnosed with bronchial stenosis through bronchoscopy. A comprehensive flow diagram of the diagnostic approach for these 24 pediatric patients is shown in Figure 2.
| NO; Gene; Nucleotide Change | Amino Acid Change | Pathogenicity Rating | Diagnosis | Genotype |
|---|---|---|---|---|
| 1 | ||||
| CFTR | CF | Compound heterozygous | ||
| c.1766 + 5G > T | - | PS3 + PM2 + PM3 + PP3 | ||
| c.263T > G | p.Leu88X | PVS1 + PM2 + PM3 | ||
| 2 | ||||
| CFTR | ||||
| c.1766 + 5G > T | - | PS3 + PM2 + PM3 + PP3 | CF | Homozygous |
| 4 | ||||
| DNAH5 | PCD | Compound heterozygous | ||
| c.4314delT | p.Asn14 38Lysfs*10 | PVS1 + PM2 | ||
| c.877dupA | p.Arg293Lysfs*6 | PVS1 + PM2 | ||
| 5 | ||||
| DNAH11 | PCD | Compound heterozygous | ||
| c.3426 - 1G > A | - | PVS1 + PM2 | ||
| c.5460 + 5G > C | - | PM2 | ||
| 6 | ||||
| RSPH4A | PCD | Compound heterozygous | ||
| c.1774-c.1775delTT | p.Leu592fsTer 5 | PVS1 + PM2 | ||
| c.1949A > G | p.His650Arg | PM2 + PP3 | ||
| 7 | ||||
| DNAH11 | PCD | Compound heterozygous | ||
| c.117499-c.11752delGTTA | p.Val3917fs Ter20 | PVS1 + PM2 | ||
| c.5822G > C | p.Trp1941Ser | PM2 + PP3 | ||
| 8 | ||||
| BTK | ||||
| c.1631 + 5G > T | - | PM2 + PP3 | XLA | Hemizygous |
| 9 | ||||
| ATM | ||||
| c.6397C > T | p.Gln2133 Ter,924 | PVS1 + PM2 | A-T | Homozygous |
| 10 | ||||
| IL2RG | ||||
| c.759G > T | p.Glu253Asp | PS + PM1 + PP3 | X-SCID | Hemizygous |
| 22 | ||||
| LRBA | Common variable immunodeficiency-8 with autoimmunity | Compound heterozygous | ||
| c.7888G > A | p.Val2630Ile | PM2 + PP3 | ||
| c.649A > G | p.Ile217Val | PM2 + PP3 | ||
| 23 | ||||
| MCM10 | ||||
| c.2119+18T>G | - | PM3 + PP3 | Immunodeficiency-80 | Homozygous |
Positive Genetic Results of 11 Children with Challenging-to-Diagnose Respiratory Diseases
| Disease Category | N | Key Phenotypic Features | Genotype-Phenotype Correlation |
|---|---|---|---|
| CF (n = 2) | |||
| CFTR compound heterozygous | 1 | Bronchiectasis, pancreatic insufficiency, malnutrition | Classic CF phenotype |
| CFTR homozygous | 1 | Severe bronchiectasis, Pseudomonas colonization | Severe CF phenotype |
| PCD (n = 4) | |||
| DNAH5 mutations | 2 | Situs inversus (50%), chronic wet cough (100%) | Typical PCD phenotype |
| DNAH11 mutations | 1 | Bronchiectasis, chronic rhinosinusitis | Mild PCD phenotype |
| RSPH4A mutations | 1 | Neonatal respiratory distress, bronchiectasis | Early-onset PCD |
| Immunodeficiencies (n = 5) | |||
| BTK mutation | 1 | Recurrent bacterial infections, hypogammaglobulinemia | Classic XLA |
| ATM mutation | 1 | Ataxia, immunodeficiency, growth delay | A-T syndrome |
| IL2RG mutation | 1 | Infections, lymphopenia | X-SCID |
| LRBA mutations | 1 | Autoimmunity, chronic diarrhea | CVID-like |
| MCM10 mutations | 1 | Infection, lymphopenia, Thymus hypoplasia, pericardial effusion | Untypical immunodeficiency |
Genotype-Phenotype Correlations by Disease Category

A comprehensive flow diagram of diagnostic approach for 24 pediatric patients with difficult-to-diagnose respiratory diseases
5. Discussion
In this study, 11 cases (45.83%) were identified to have monogenic diseases. The diseases diagnosed here include PCD, CF, and immunodeficiency-associated diseases. Similarly, a study conducted at Beijing Children's Hospital (4), involving 107 children, identified 51 genetic variants in 37 patients, resulting in a 34.6% diagnostic rate; the most common diagnoses were primary immunodeficiency disease (18 cases) and PCD (9 cases).
Although phenotypic analysis is a key diagnostic step that should be conducted prior to genetic analysis, heterogeneous disease characteristics associated with certain respiratory diseases generally make it difficult to comprehensively describe phenotypes for such diseases. In our study, the predominant manifestations included cough (91.67%) and wheezing (41.67%). In the aforementioned study (4), 21.6% of patients with no clinical diagnosis due to nonspecific phenotypes obtained definitive diagnoses through genetic testing. Compared to the results obtained here for 14 bronchiectasis cases, 5 cases (58.33%) tested positive for inherited diseases. Of note, early and accurate diagnosis is critically important to obtain positive treatment outcomes for bronchiectasis (6).
While it was previously believed that CF was very rare in China, here, 2 children were diagnosed with CF. This is consistent with recent results indicating that CF is not so rare in China (7, 8). Notably, the disease characteristics of Chinese CF pediatric patients include bronchiectasis (95.2%) and allergic bronchopulmonary aspergillosis (42.6%), with predominant airway organisms including P. aeruginosa (66.7%) and S. aureus (33.7%). Nevertheless, neonatal screening for CF and sweat tests to diagnose CF are not widely available in China (9). This forces clinicians to rely on phenotyping and genetic testing to diagnose CF. Importantly, 2 CF patients harbored the CFTR c.1766 + 5G > T variant, which has been reported to be the most frequently observed variant in southern and eastern coastal regions of China (10, 11). The other 3 children with single CFTR mutations shared a phenotype characterized by recurrent wheezing and excessive sputum production after infection, which indicates that CFTR may be related to asthma. It has also been reported that asthma patients carrying single CFTR mutations exhibit mild CF symptoms associated with abundant airway accumulation of mucus in addition to the asthma phenotype (12).
Most patients in this study were diagnosed with PCD (5/24). The diagnoses were based on 4 key clinical PCD markers: Neonatal respiratory distress of unknown etiology; chronic wet cough beginning before 6 months of age; chronic daily nasal congestion beginning before 6 months of age; and organ left/right asymmetry. Patients should be suspected of having PCD if they possess at least 2 of the 4 above-mentioned features to improve diagnostic specificity and sensitivity (13). Children with PCD often cough up a noticeable amount of mucus from an early age, with symptoms improving somewhat but not completely disappearing after weather changes or standard asthma or anti-allergic treatments (14). Furthermore, transmission electron microscopy (TEM) studies have shown that about 20 - 30% of ciliary structures of PCD patients are normal (15). In hospitals with limited resources, genetic testing, TEM, or nasal nitric oxide (nNO) may not be available; thus, phenotypes should be carefully evaluated to distinguish CF from PCD.
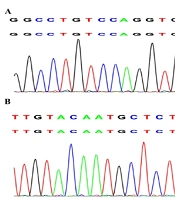
Five children in this study were diagnosed with immunodeficiencies. Patient 8 experienced a recurrent wet cough for over 10 years accompanied by bronchiectasis, sinusitis, otitis media, and low IgG, IgA, and IgM levels, consistent with an XLA phenotype. The novel BTK splice site variant c.1631 + 5G > T (Figure 3A) affects the canonical GT splice donor sequence and is predicted to cause exon skipping and premature termination. Most children with XLA are diagnosed primarily based on the presence of bronchopneumonia, bronchiectasis, chronic obstructive pulmonary emphysema, asthma, pulmonary abscess, empyema, chronic pleuritis, atelectasis, and tuberculosis (16). Patient 8 exhibited a milder phenotype with recurrent respiratory infections and reduced yet detectable B-cell counts. This variation might be due to the mutation's specific position, which allows some residual kinase activity, as seen in other BTK variants in the same domain (17). By contrast, AT is characterized by progressive cerebellar ataxia, conjunctival and skin telangiectasia, immune deficiency, recurrent paranasal sinusitis, and pulmonary infection in infancy caused by serious nervous system damage (18). Here, patient 9 was homozygous for the c.6397C > T mutation (Figure 3B) in the ATM gene. She experienced recurrent respiratory infections and exhibited a markedly decreased IgA level. Moreover, the patient exhibited poor neurological development and experienced multiple febrile convulsions. Further, physical examinations showed she had ataxia without telangiectasia, aligning with findings indicating that only some ataxic patients show telangiectasia signs (19). Moreover, X-SCID is a rare immunodeficiency disease caused by mutation of the gene encoding the common gamma chain (γC), namely IL2RG. Typical manifestations include weight loss, chronic candida infection, and chronic diarrhea that is often accompanied by conditional pathogen or viral infections; however, atypical patients may exhibit signs of relatively milder immunodeficiencies (20). Here, patient 10 harbored an IL2RG gene c.759G>T mutation; this is a relatively benign variant that has no apparent effect on the overall lymphocyte number, relative proportions of immune T-cells, B-cells, and NK cells, or serum immunoglobulin concentration. Meanwhile, patient 22 had presumed compound heterozygous mutations of the LRBA gene, including the reported c.7888G>A mutation and the novel c. 649A > G mutation, and exhibited a recurrent wet cough, pneumonia, hemoptysis, bronchiectasis, and severe malnutrition. Furthermore, immunodeficiency-80 is an autosomal recessive immunologic disorder with variable manifestations. Baxley et al. (21) revealed that MCM10 function must exceed a certain threshold to support normal tissue cell lineage-specific development. The diagnosis of patient 23 was confirmed through genetic detection of a novel homozygous c.2119 + 18T > G MCM10 gene mutation as the potential cause of the observed phenotype. MCM10 has not been previously associated with respiratory disorders. The variant c.2119 + 18T > G affects the canonical TG splice donor sequence and is predicted to cause exon skipping and premature termination. Disruption of this interaction could impair DNA replication fidelity, potentially leading to genomic instability in highly proliferative tissues such as the respiratory epithelium.
; B, ATM c.6397C > T mutation (case 9)")
Gene sequencing results obtained for some of the children: A, BTK c.1631 + 5G > T mutation (case 8); B, ATM c.6397C > T mutation (case 9)
In patients with challenging-to-diagnose respiratory diseases, a systematic diagnostic approach can speed up treatment planning. Depending on the clinical history and the severity of the disease, multiple diagnostic methods might be necessary. Initial tests typically include radiographic and laboratory studies, with further histologic and functional tests used to refine the diagnosis. The HRCT is useful for identifying specific lung abnormalities. Early use of specialized laboratory tests, including complete blood count, immune globulin or lymphocyte panel, sweat test, and nNO, could help definitively diagnose the underlying disease. Microbiologic evaluations are important for identifying infections or bronchiectasis exacerbations. Lung function tests should be used to monitor changes in symptoms. However, considering the limitations of testing devices and the lack of local clinical expertise, genomic testing may be more cost‐effective and time‐efficient.
There are several limitations of this research that should be noted. First, this study lacked a control group with similar symptoms but confirmed non-genetic etiology. This limits our ability to determine the specificity of the clinical features associated with particular genetic variants. Second, the study sample was relatively small. This limits the power of comparative analyses and may affect the reliability of the calculated diagnostic values. Third, this study was conducted in a single center and lacked sweat testing and nasal exhaled nNO testing. Fourth, technical limitations related to data analysis may have affected the final results. Thus, studies with larger sample sizes, multi-center longitudinal phenotypic mining, and genetic analyses should be conducted to improve diagnostic rates in the future. Despite cost and infrastructure challenges, especially in resource-limited areas, targeted testing in high-risk patients can enhance diagnosis and resource use. As sequencing costs continue to decrease and interpretation improves, wider implementation is expected to become more cost-effective and feasible.
In summary, phenotypic analysis and genetic testing are important for identifying pediatric challenging-to-diagnose respiratory diseases, especially cases with suspected CF, PCD, and immunodeficiencies. A systematic, strategic, phenotype-guided genetic testing approach can enhance diagnosis, management, and outcomes. Future research should involve larger sample sizes, multi-center studies, and functional validation with follow-up on variants of unclear significance.
