
Fulltext
Kabuki make-up syndrome (KMS) is a rare multiple congenital anomalies/mental retardation syndrome of unknown cause, first described independently by Niikawa and Kuroki. It is characterized by distinctive facial features, skeletal anomalies, dermatoglyphic abnormalities, short stature, and mental retardation. A group of abnormalities with involvement of other organs can lead to diagnosis of KMS.
The authors report a 4-and-half-year old girl with KMS, admitted for bilateral dislocation of the hip. On this case, we try to explain the syndrome and to allow pediatricians and other physicians to become familiar with this syndrome.
KMS (OMIM#147920) is a rare dysmorphic syndrome that was first described in 1981 by Niikawa et al[1] and Kuroki et al[2]. The name of the syndrome, Kabuki make-up, refers to resemblance of the facial traits of KMS patients to the make-up of actors in Kabuki, the traditional Japanese theatre[1,2].
KMS is more common in Japan, although it has been reported from different geographic regions in the world[3]. Only few hundred cases have been described, and it is expressed as 1/32000 incidence in Japan[4]. We recently observed a 4 and half year old girl with typical KMS presented with bilateral dislocation of the hip.
The patient was referred to pediatric orthopedic department for bilateral dislocation of the hip. Her parents were healthy with no consanguinity.
Psychomotor development was delayed with speech defects. On physical examination at the age of 4 years, the patient’s height was 92 cm, weight 15 kg. She had charcteristic facial dysmorphism with high arched eyebrows that were sparse in the lateral part, long palpebral fissures, prominent and protruding ears and lip pits on the lower lip, depressed nasal tip (Fig. 1A). She also had joint laxity, brachydactyly V (Fig. 1B) and finger tip pads (Fig. 1C).
Other findings were normal. Chest radiography showed kyphotic deformity of the spine. Echocardiography was normal. Abdomen and pelvic sonography showed pelvic situation of the right kidney. Pelvic radiography showed bilateral dislocation of the hip (Fig. 2). She was diagnosed as Kabuki make-up syndrome due to clinical findings.
She was operated for bilateral dislocation of the hip with, in the first, the right hip by open reduction, iliopsoas and adductor releases, capsuloraphy and Salter innominate osteotomy, and the same technique in the left hip with good results.
KMS occurs mostly sporadic, although some familial cases have been reported[6]. Inheritance
The authors report a 4-and-half-year old girl with KMS, admitted for bilateral dislocation of the hip. On this case, we try to explain the syndrome and to allow pediatricians and other physicians to become familiar with this syndrome.
KMS (OMIM#147920) is a rare dysmorphic syndrome that was first described in 1981 by Niikawa et al[1] and Kuroki et al[2]. The name of the syndrome, Kabuki make-up, refers to resemblance of the facial traits of KMS patients to the make-up of actors in Kabuki, the traditional Japanese theatre[1,2].
KMS is more common in Japan, although it has been reported from different geographic regions in the world[3]. Only few hundred cases have been described, and it is expressed as 1/32000 incidence in Japan[4]. We recently observed a 4 and half year old girl with typical KMS presented with bilateral dislocation of the hip.
The patient was referred to pediatric orthopedic department for bilateral dislocation of the hip. Her parents were healthy with no consanguinity.
Psychomotor development was delayed with speech defects. On physical examination at the age of 4 years, the patient’s height was 92 cm, weight 15 kg. She had charcteristic facial dysmorphism with high arched eyebrows that were sparse in the lateral part, long palpebral fissures, prominent and protruding ears and lip pits on the lower lip, depressed nasal tip (Fig. 1A). She also had joint laxity, brachydactyly V (Fig. 1B) and finger tip pads (Fig. 1C).
Other findings were normal. Chest radiography showed kyphotic deformity of the spine. Echocardiography was normal. Abdomen and pelvic sonography showed pelvic situation of the right kidney. Pelvic radiography showed bilateral dislocation of the hip (Fig. 2). She was diagnosed as Kabuki make-up syndrome due to clinical findings.
She was operated for bilateral dislocation of the hip with, in the first, the right hip by open reduction, iliopsoas and adductor releases, capsuloraphy and Salter innominate osteotomy, and the same technique in the left hip with good results.
KMS occurs mostly sporadic, although some familial cases have been reported[6]. Inheritance
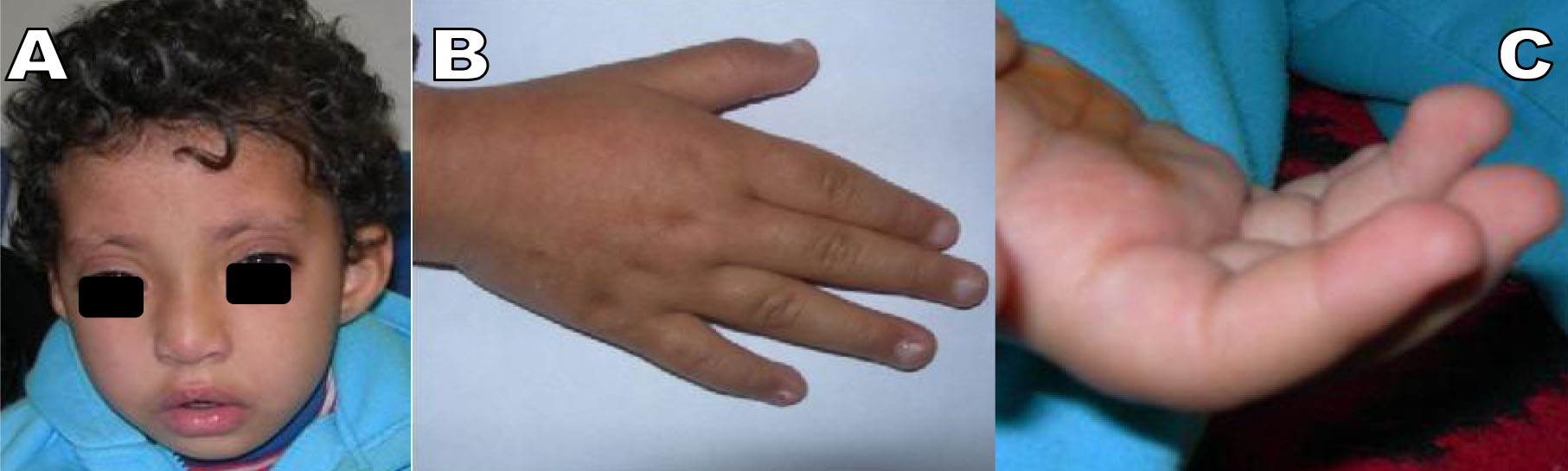
Fig. 1: A) Characteristic facial dysmorphism with high arched eyebrows that were sparse in the lateral part, long palpebral fissures, prominent and protruding ears and lip pits on the lower lip, depressed nasal tip. B): Brachydactyly V and C) finger tip pads in patient with Kabuki make-up syndrome.
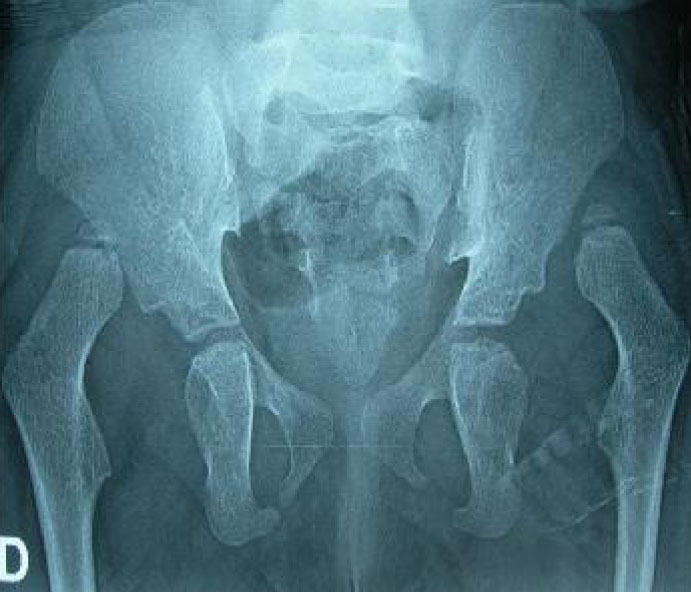
Fig. 2: Pelvic radiography shows bilateral dislocation of the hip
is thought to be autosomal dominant on X-linked recessive; although several cases with KMS features have been reported with different chromosomal anomalies, none have had an autosomal cytogenetic aberration in common[5,6].
In 1988, Niikawa et al[7] reported 62 patients diagnosed with KMS. Based on the findings in these patients, five cardinal manifestations were defined. These included a peculiar face characterized by eversion of the lower lateral eyelid, arched eyebrows with the lateral one-third dispersed or sparse, depressed nasal tip, and prominent ears in 100â , skeletal anomalies characterized by deformed spinal column with or without sagittal cleft vertebrae, and brachydactyly V in 92â , dermatoglyphic abnormalities including fingertip pads, absence of digital triradius c and/or d, and increased digital ulnar loop and hypothenar loop patterns in 93â , mild to moderate mental retardation in 92â , and postnatal growth deficiency in 83â of their patients. For positive diagnosis, it is necessary to have minimally peculiar facial appearance, mental retardation and postnatal growth deficiency[7-10]. Our patient had a typical KMS. Philip et al[11] have commented from various European centers that skeletal abnormalities are common in the syndrome, but that they are usually not specific radiologically. They included shortening of metacarpals and phalanges in the majority of cases, syndactyly and joint laxity, short stature, scoliosis and short fifth fingers[10].
The high risk of dislocation of the hip has been attributed to the generalized joint laxity that was observed in 10 of 16 cases[8,9].
Structural heart defects are encountered in 32â -58â of children, and non specific congenital heart defect predominates[7,9].
Neonatal hypotonia and feeding difficulties, recurrent infections, reno-urinary malformations, diaphragmatic anomalies, neurological symptoms, abnormalities of the central nervous system, obesity, and precocious puberty are also reported in this syndrome[9,10].
