
Fulltext
Among small blue round cell tumors, primitive neuroectodermal tumors (PNETs) are a group of Ewing sarcoma family of tumors that are highly aggressive, poorly differentiated and form a group of tumors defined by their appearance that are thought to derive from postganglionic parasympathetic primordial cells located throughout the parasympathetic autonomic nervous system. Batsakis et al divided PNET family of tumors into the following 3 groups based on the tissue of origin: peripheral PNET, CNS PNET and neuroblastoma[1]. The peripheral PNET (pPNET) is now thought to be identical to Ewing family of tumors (EFTs). Tumors that demonstrate neural differentiation by light microscopy, immunohistochemistry, or electron microscopy have been traditionally called PNETs, and those that are undifferentiated by these analyses have been diagnosed as Ewing's sarcoma[2]. Among the cPNET's are medulloblastoma, pineoblastoma, cerebral neuroblastoma, ependymoblastoma, medulloepithelioma, primary rhabdomyosarcoma, and atypical teratoid/rhabdoid tumor[2].
Although these tumors are exceedingly rare, the annual incidence of tumors from birth to age 20 years is 2.9 per million population. In most large series published to date, pPNETs usually present in the second decade of life, with a slight male predilection. They account for 4-17% of all pediatric soft tissue tumors[3].
Our case was an 11 year old girl with fever and severe pain in lower extremities 1 week before admission. She complained of severe abdominal pain since two weeks prior to admission. At admission body temperature was febrile and blood pressure normal. Physical examination revealed no erythema, swelling or decreased range of motion in lower extremities joints but the patient was suffering from severe pain in both extremities with no tolerance of weight bearing. Abdominal ultrasound showed hepatospleno-megaly and adenopathy. Initial laboratory tests showed normal leukocyte count (65% neutrophils), Hb was 9.5g/dL and platelet count was 757000/mm3. Liver enzymes were normal but alkaline phosphatase was 526U/L. Erythrocyte sedimentation rate was 105 and C-reactive protein was highly positive (174mg/l). Other investigation including Wright, Coombs Wright and 2ME, FANA, Anti dsDNA, cANCA and pANCA were negative. Urinalysis, urine and blood culture were negative.
Chest X-ray revealed a massive right sided pleural effusion associated with a large underlying mass (Fig 1). In thoracic CT scan with IV contrast, there was a large heterogeneously enhancing mass measured 78×50mm in right hemithorax with pleural effusion, 4th and 5th rib involvement and periosteal reaction (Fig. 2). There were also two other small solid nodules in semilateral hemithorax and two nodules in peripleural fat. No adenopathy was detected in mediastinum or axillary regions. Abdominal CT scan was unremarkable. Bone marrow aspiration showed hyper cellularity without evidence of malignant cells. Whole body radioisotope scan showed increased radiotracer uptake in posterior arc of one of the right middle ribs and along the upper two thirds of both tibiae. Mild irregular uptake was also noted along both femurs. Ultrasound guided mass biopsy showed cellular aggregates of small round cells and strands of fibrous tissue and tumor cells expressed CD99,neuron-specific enolase, S100 protein and chromogranin by immunohistochemistry study.Diagnosed as PENET the patient was treated by systemic chemotherapy and localized radiotherapy. The patient had no evidence of pain after 5 days and no evidence of disease after 5 months of treatment.
PNETs, described originally by Askin and colleagues, are infrequent thoracic tumors found in infants and children[4]. The histogenesis of this tumor remains uncertain and is suspected to arise from the intercoastal nerves[5]. Frequently, this entity appears as a chest wall mass, with rapid growth that may involve the pleura[6]. A similar chromosomal translocation t(11;22)(q24;q12)
Our case was an 11 year old girl with fever and severe pain in lower extremities 1 week before admission. She complained of severe abdominal pain since two weeks prior to admission. At admission body temperature was febrile and blood pressure normal. Physical examination revealed no erythema, swelling or decreased range of motion in lower extremities joints but the patient was suffering from severe pain in both extremities with no tolerance of weight bearing. Abdominal ultrasound showed hepatospleno-megaly and adenopathy. Initial laboratory tests showed normal leukocyte count (65% neutrophils), Hb was 9.5g/dL and platelet count was 757000/mm3. Liver enzymes were normal but alkaline phosphatase was 526U/L. Erythrocyte sedimentation rate was 105 and C-reactive protein was highly positive (174mg/l). Other investigation including Wright, Coombs Wright and 2ME, FANA, Anti dsDNA, cANCA and pANCA were negative. Urinalysis, urine and blood culture were negative.
Chest X-ray revealed a massive right sided pleural effusion associated with a large underlying mass (Fig 1). In thoracic CT scan with IV contrast, there was a large heterogeneously enhancing mass measured 78×50mm in right hemithorax with pleural effusion, 4th and 5th rib involvement and periosteal reaction (Fig. 2). There were also two other small solid nodules in semilateral hemithorax and two nodules in peripleural fat. No adenopathy was detected in mediastinum or axillary regions. Abdominal CT scan was unremarkable. Bone marrow aspiration showed hyper cellularity without evidence of malignant cells. Whole body radioisotope scan showed increased radiotracer uptake in posterior arc of one of the right middle ribs and along the upper two thirds of both tibiae. Mild irregular uptake was also noted along both femurs. Ultrasound guided mass biopsy showed cellular aggregates of small round cells and strands of fibrous tissue and tumor cells expressed CD99,neuron-specific enolase, S100 protein and chromogranin by immunohistochemistry study.Diagnosed as PENET the patient was treated by systemic chemotherapy and localized radiotherapy. The patient had no evidence of pain after 5 days and no evidence of disease after 5 months of treatment.
PNETs, described originally by Askin and colleagues, are infrequent thoracic tumors found in infants and children[4]. The histogenesis of this tumor remains uncertain and is suspected to arise from the intercoastal nerves[5]. Frequently, this entity appears as a chest wall mass, with rapid growth that may involve the pleura[6]. A similar chromosomal translocation t(11;22)(q24;q12)

Fig. 1: Massive right sided pleural effusion associated with a large underlying mass in Chest X-ray in
occurring in both Ewing's sarcoma and PNET lesions suggest that these tumors are closely related[7,8]. In the thoracic area, these tumors are invasive and prone to destroying bone, invading the retroperitoneal space, and spreading to lymph nodes, adrenals, and liver [7].
The treatment of PNET involves combined modality therapy with chemotherapy and local therapy offered by surgical resection, radiation or both[8]. The 5-year relative survival estimates (based on data from 2001–2006) are 64% in children (1–9 years) and 35% in adults (20+ years)[9].
Shamberger et al treated 21 infants and children with malignant tumors of the chest wall from 1976 to 1989, of which fifteen were classified as Askin's tumors or Ewing's sarcoma. Ten patients with localized disease received combined therapy including surgical resection, radiotherapy, and chemotherapy, six of them were disease-free 3.5 to 9 years (median, 5 years) following diagnosis[10].
Our case is an example of childhood malignancies with predominantly musculoskeletal symptoms. She was admitted at first with diagnosis of juvenile idiopathic arthritis because of severe bone pain in lower extremities that led to intolerance of weight bearing and severe child irritability. Although leukemia and lymphoma are the most common cancers accompanied with musculoskeletal disorders in children, other solid tumors such as neuroblastoma and PNETs should be considered in differential diagnosis in any child presenting with bone symptoms especially bone
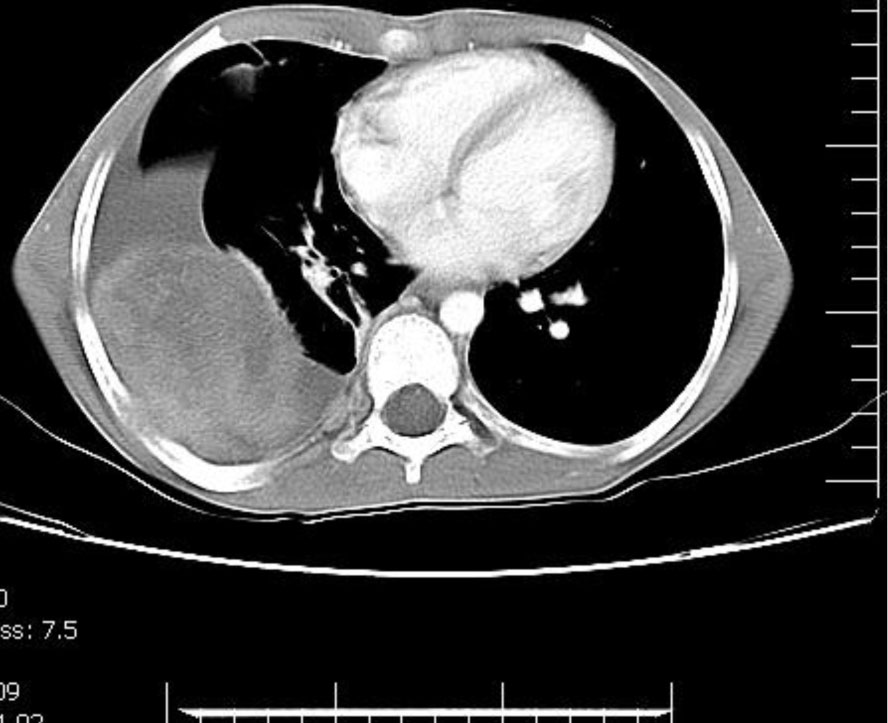
Fig. 2: A large heterogeneously enhancing mass in right hemithorax with pleural effusion, rib involvement and periosteal reaction
presenting with bone symptoms especially bone pain.
occurring in both Ewing's sarcoma and PNET lesions suggest that these tumors are closely related[7,8]. In the thoracic area, these tumors are invasive and prone to destroying bone, invading the retroperitoneal space, and spreading to lymph nodes, adrenals, and liver [7].
The treatment of PNET involves combined modality therapy with chemotherapy and local therapy offered by surgical resection, radiation or both[8]. The 5-year relative survival estimates (based on data from 2001–2006) are 64% in children (1–9 years) and 35% in adults (20+ years)[9].
Shamberger et al treated 21 infants and children with malignant tumors of the chest wall from 1976 to 1989, of which fifteen were classified as Askin's tumors or Ewing's sarcoma. Ten patients with localized disease received combined therapy including surgical resection, radiotherapy, and chemotherapy, six of them were disease-free 3.5 to 9 years (median, 5 years) following diagnosis[10].
Our case is an example of childhood malignancies with predominantly musculoskeletal symptoms. She was admitted at first with diagnosis of juvenile idiopathic arthritis because of severe bone pain in lower extremities that led to intolerance of weight bearing and severe child irritability. Although leukemia and lymphoma are the most common cancers accompanied with musculoskeletal disorders in children, other solid tumors such as neuroblastoma and PNETs should be considered in differential diagnosis in any child presenting with bone symptoms especially bone
Fig. 2: A large heterogeneously enhancing mass in right hemithorax with pleural effusion, rib involvement and periosteal reaction
presenting with bone symptoms especially bone pain.