




1. Introduction
Pyruvate dehydrogenase complex deficiency (PDHD) is a genetic mitochondrial disorder. Pyruvate dehydrogenase complex (PDC) is the key rate-limiting enzyme for glucose oxidation (1). Although a congenital deficiency of the PDC has been reported for the first time more than 40 years ago, the incidence and prevalence of this remain unknown due to lethality (2).
The clinical manifestations of PDHD are very various from lactic acidosis in newborn period with early lethality to chronic neurodegenerative condition. Progressive neurological symptoms usually start in infancy, but may begin at birth or in later childhood (3). They may include developmental delay, hypotonia, seizures, microcephaly, facial dysmorphism, ataxia, nystagmus and Leigh-like syndrome (4).
In the past, 1-14C-pyruvate-based assay of PDC in blood lymphocytes, cultured fibroblasts or skeletal muscles were used for diagnosis of PDHD. With the development of next-generation sequencing (NGS) technique, genetic testing is also being used in the diagnosis of PDHD (5). Nevertheless, it is difficult to make a diagnosis of PDHD in neonatal period. Here, we report a preterm infant with neonatal onset PDHD who presented with persistent lactic acidosis, scoliosis, polydactyly and agenesis of corpus callosum.
2. Case Presentation
A male infant was born at 31 weeks and 3 days of gestational age with a birth weight of 1,310 g (16th percentile) via cesarean section due to maternal preeclampsia. There was no initial crying at birth and initial heart rate was below 100 beats/min, so positive pressure ventilation was administered. At 2 minutes, after the immediate endotracheal intubation, effective ventilation was achieved. The Apgar score was 3 and 6 at 1 and 5 minutes after birth, respectively. The baby was admitted to the neonatal intensive care unit (NICU).
The baby’s mother was 39 years old, G1P1. She was diagnosed with schizophrenia and diabetes mellitus and she had been treated with dopamine stabilizer, selective serotonin reabsorption inhibitor and insulin. She had not received antenatal care before 26 weeks of gestation. Prenatal ultrasonography was performed at 26 weeks of gestation, and revealed scoliosis, agenesis of corpus callosum, hemivertebra and intrauterine growth retardation (IUGR).
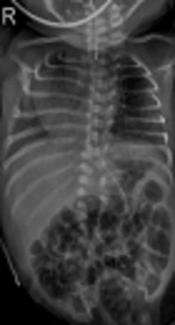
At birth, he was flaccid and apneic. Physical examination identified scoliosis and polydactyly under left thumb (Figure 1). His head circumference was 28.5 cm (40th percentile).
; B, clinical photograph shows polydactyly under left thumb; C, cranial ultrasound shows corpus callosum agenesis and increased parenchymal echogenicity (on 3rd day of life)")
A, plain radiograph shows scoliosis and vertebral anomalies (at birth); B, clinical photograph shows polydactyly under left thumb; C, cranial ultrasound shows corpus callosum agenesis and increased parenchymal echogenicity (on 3rd day of life)
On the day of birth, his laboratory results were as follows: A biochemical analysis showed severe metabolic acidosis (venous pH: 6.96, pCO2: 52.2 mmHg, base excess: -20.1). Laboratory tests revealed a white blood cell count of 10,960/mm3, hemoglobin level of 9.8 g/dL, platelet count of 264,000/mm3, and the C-reactive protein level was < 0.03 mg/L. The serum eletrolytes were sodium 134.1 mEq/L, potassium 4.88 mEq/L, chloride 103 mEq/L, calcium 7.8 mg/L and the blood glucose was 127 mg/dL. Elevated lactate level in blood (20.66 mmol/L; reference values [RV] 0.7 - 2.1 mmol/L) and pyruvate level (1.63 mmol/L; RV 0.01 - 0.10 mmol/L), with a normal lactate-to-pyruvate ratio, were found. Elevated lactate level in cerebrospinal fluid (14.87 mmol/L; RV 0.7 - 2.1 mmol/L) and pyruvate level (1.49 mmol/L; RV 0.03 - 0.15 mmol/L) with a normal lactate-to-pyruvate ratio, was also demonstrated. Viral and bacterial tests were negative. Plasma amino acids showed increased concentrations of alanine (1773.8 µmol/L; RV 167 - 439 µmol/L) and normal level of leucine (57.1 mmol/L; RV 79 - 150 mmol/L), isoleucine (16.1 mmol/L; RV 40 - 90 mmol/L) and alloisoleucine (not detected; RV not detected), urine organic acid analysis revealed a high excretion of lactate (4864.6 mmol/mol creatinine; RV < 270 mmol/mol creatinine). A cranial ultrasound detected corpus callosum agenesis and increased parenchymal echogenicity (Figure 1). There was no renal or cardiac dysfunction. Also, there were no congenital abnormalities of the heart or abdominal organs according to ultrasonography.
Genetic testing was performed to make a diagnosis using the TruSight One Sequencing Panel kit (Illumina, San Diego, CA, USA) includes genes from the Human Gene Mutation Database (www.hgmd.cf.ac.uk/ac/index.php) and the Online Mendelian Inheritance in Man database (www.genetests.org). This targeted NGS panel revealed a hemizygous variant on the gene coding for the PDHA1 (Figure 2). Sanger sequencing confirmed a novel PDHA1 mutation (NM-000284.3:c.1157-1162delTTAAGT (p.Phe386Lys387del) on the X chromosome. The parents and sister of the patient were analyzed for the found variant using Sanger sequencing. On the familial study, the same mutation of the PDHA1 was also found in patient’s unaffected mother.
.")
Sequencing analysis of the PDHA1 gene shows an inframe deletion mutation: c.1157-1162delTTAAGT (p.Phe386Lys387del).
After confirming the diagnosis of PDHD, he was treated with thiamine (50 mg/kg/d), a ketogenic diet (ratio of fats to carbohydrates and protein was 3:1), sodium bicarbonate as needed and conservative management. After treatment started, lactate values declined to near normal levels (< 2 mmol/L) but failed to alter the devastating clinical course. The patient was found dead at 5 months of life. The cause of death was respiratory infection.
3. Discussion
The PDC is located in the mitochondrial matrix and catalyzes the production of acetyl-CoA from pyruvate produced by glycolysis (1). It consists of multiple copies of three catalytic enzymes, pyruvate dehydrogenase (E1), dihydrolipoamide transacetylase (E2) and dihydrolipoamide dehydrogenase (E3), as well as an E3 binding protein (BP) (4). There are six forms of PDHD, depending on the genetic background and damaged subunit of the enzyme complex (3). The X-linked PDHA1 gene is responsible for the majority of cases (6). The PDHA1 gene, encoding the E1α subunit is located on chromosome Xp22.1 and comprises eleven exons. Hemizygous males are generally symptomatic, whereas heterozygous females present variable expression of the mutant (7). In this case, PDHA1 mutation was inherited from patient’s unaffected mother. The patient showed severe metabolic acidosis from birth, but his mother showed normal intelligence with normal lactate and pyruvate level in blood. To date, at least 337 patients with PDHA1 mutations have been reported (8, 9). Most of them are missense changes, whereas deletion or duplication of the PDHA1 gene is rare (20% of cases) (6). In this report, we found a PDHD-causing molecular variant with a six-nucleotide deletion from the 1157th to 1162th coding nucleotide in PDHA1 gene. Although we did not conduct the enzymatic testing for PDC activity, PDC activity in lymphocyte with the same mutation in previous studies was low (53%) (1). With regard to the findings of Robinson et al. (10), showing that residual activity of the PDC ranged from 1.6% to 69.5% of controls, 53% of PDC activity is enough to say that the enzyme activity has decreased. As described above, this mutation can be classified as a “Likely pathogenic” according to the American College of Medical Genetics and Genomics (ACMG) guidelines. Additionally, a few cases are known to result from mutations in genes encoding subunits: E1β (PDHB), E2 (PDHA2), E3 (DLD), and E3BP (PDHX) or PDH phosphatase (PDP1) (3). Involvement of the E2 fraction or the E3BP protein, which is transmitted in autosomal recessive fashion, is rarer (11, 12).
The phenotypic spectrum of PDHD varies widely from severe lactic acidosis in neonatal period to progressive neurological and neuromuscular degeneration. There are three phenotypes in PDHD with a PDHA1 gene mutation: (1) neonatal onset encephalopathy with congenital lactic acidosis, prenatal brain lesions such as corpus callosum agenesis and facial dysmorphism, (2) learning disability and basal ganglia necrosis similar to Leigh syndrome and (3) relapsing episodes of weakness and ataxia with prolonged survival (4). Recognition of PDHD is difficult due to its wide variety of clinical presentations. Furthermore, nonspecific signs including poor feeding, lethargy, hypotonia, and respiratory distress make it more difficult to diagnose PDHD when it presents during the neonatal period.
PDHD is considered one of the most common biochemically proven causes of lactic acidosis (13). In neonates, various causes of metabolic acidosis include: Birth asphyxia, cold stress, hypovolemia, sepsis, congenital heart disease, renal disease, and inborn errors of metabolism. Especially preterm infants are more susceptible to metabolic acidosis because they are more under the disorders that cause metabolic acidosis such as cold stress, infection, respiratory distress syndrome and immature renal function. When metabolic acidosis is unexplained, persistent, and severe in preterm infants, PDHD should be considered and further evaluation including blood lactate, pyruvate, ammonia, plasma amino acid, and urine organic acid should be cheked (14).
Structural brain abnormalities including ventriculomegaly and agenesis, dysgenesis, or hypolasia of the corpus callosum have long been noted as a common feature of PDHD (4). The key role of PDC in providing energy necessary for neuronal migration makes the brain highly sensitive to PDHD. Prenatal energy failure may account for structural brain abnormalities (1). In contrast, skeletal abnormalities of PDHD are poorly known. Fujii et al. (15) described a patient with PDHD who presented with left thumb malposition and scoliosis.
Recently, widespread availability of genetic testing has resulted in an increasing awareness of the new monogenic disorders (16). NGS technique has revealed a significant number of genetic defects. In the past, PDHD was diagnosed from PDC activity measured by decarboxylation of 1-14C-pyruvate to 14CO2 (4). As was demonstrated in this case, NGS technique was useful for making a diagnosis of PDHD. Furthermore, whole-exome sequencing (WES) could be helpful for the rapid and simultaneous screening of candidate variants and genes linked to PDHD in neonates.
PDHD is a rare and fatal disease in case of neonatal onset. Therefore, prompt and exact diagnosis of PDHD has implications for clinical management in neonates. When a severe and persistent lactic acidosis is noted at birth, PDHD should be suspected and evaluated using molecular analysis because of progression and lethality. We recommend targeted NGS panel or WES in neonates with clinical and biochemical features of PDHD. WES which enables detection of variants in a wide range of known genes as well as in mutations in other genes is becoming the method of choice for the diagnostics of PDHD.
3.1. Conclusions
In conclusion, PDHD should be considered among the differential diagnosis in neonates who present with persistent lactic acidosis, agenesis of corpus callosum, scoliosis and polydactyly. This case demonstrates early diagnosis of PDHD in preterm infant using targeted NGS panel. Furthermore, future research should investigate the possible correlations between the genotype and phenotype with different PDHD mutations.
