




1. Background
Five cases of Pneumocystis pneumonia among homosexual males were reported on June 5, 1981, in the Centers for Disease Control's Morbidity and Mortality Weekly Report (1). That study signaled the beginning of the HIV/AIDS epidemic, which has now infected more than 75 million people (2). Acquired immunodeficiency syndrome (AIDS) is the outcome of HIV infection that has not been carefully controlled. Since the epidemic's beginning, 36.3 million people have died from AIDS-related diseases (3-5). Treatment for AIDS is complicated due to the unavailability of vaccines and the presence of viral reservoirs (6, 7). Combination antiretroviral therapy (cART) is the most common treatment for HIV/AIDS (8). The cART has turned HIV-1 infection into a chronic illness with reduced viral load and mortality (9). Drug resistance and pharmacological toxicity are the disadvantages of cART. Therefore, new treatment targets are required (10).
The HIV-1 therapy medicines are commercially available. A combination of two or more active antiretroviral medicines from different classes is advised for the most effective viral load suppression. Two nucleoside reverse transcriptase inhibitors (NRTIs) plus a third medication, such as an integrase-strand transfer inhibitor (INSTI), non-nucleoside reverse transcriptase inhibitor (NNRTI), or pharmacologically enhanced protease inhibitor (PI), are often used in the first phase of treatment (11). As a result, cART is capable of halting viral replication (12). Human immunodeficiency virus type-1 (HIV-1) and other retroviruses need the retroviral integrase enzyme for replication, which has three structurally different domains: An N-terminal domain, a catalytic core, and a C-terminal domain (13). HIV integrase is an attractive target for therapeutic administration since it lacks a functional human homolog (14). Viral integrase, a 288 amino-acid, 32 kDa enzyme, facilitates the integration of double-stranded viral DNA into the host cell genome (15). The two phases in the integration process are 3'-processing (3'-P) and strand transfer (ST). The first step is the integration of viral DNA into IN in the cytoplasm. In this reaction, IN catalyzes the cleavage of two (or three, depending on in vitro conditions) nucleotides from each of the 3' ends of the DNA (3'-processing) (16, 17). Strand transfer occurs after the preintegration complex (PIC) is transported to the nucleus of infected cells. The viral DNA's 3' hydroxyl group links to the human chromosome during the strand transfer step. The catalytic domain of integrase contains a DDE motif (D64, D116, and E152 amino acids) paired with two Mg2+ ions in the enzyme's active site, which are essential for catalytic activity (18, 19). Therefore, metal-based chelation is an approach for inhibiting strand transfer with more selectivity. Elvitegravir, dolutegravir, cabotegravir, and other FDA-approved IN inhibitor pharmacophores (Figure 1) indicate that an INSTI is designed with heteroatoms as a chelating motif and a coplanar hydrophobic aryl group (20-24).

Figure 1.
Structure of FDA-approved IN inhibitors
Finding new and safe anti-HIV agents is a top priority since all current inhibitors have been unsatisfactory against HIV strains. The HIV IN inhibitory activity of styrlquinoline and salicylhydrazide aromatics (as seen by structures 1 and 2 in Figure 2) was previously found, but clinical usefulness was limited by their apparent cytotoxicity (25-27). Therefore, a core with minimum toxicity and specific effect is required.
The 4-hydroxyquinoline scaffold found in various structures shows different biological effects, including antibacterial, antioxidant, anti-HIV, anti-fibrosis, antiviral, and anticancer activities (28-31). In addition, our group previously reported some 4-hydroxyquinoline derivatives possessing promising anti-HIV activity with no significant cytotoxicity (as exemplified by structure 3) (32, 33). This study applied the hybridization technique to obtain a new class of compounds endowed with HIV inhibitory activity and no cytotoxicity. A series of compounds (8a-g) was achieved by merging the 8-methyl-4-hydroxyquinoline group of our previously reported compound 3 with the pharmacophores of the salicylhydrazide (1). In another series of designed compounds (9a-c and 10a-f), we changed the distance between the substituted phenyl moiety and the carbohydrazide group by one and two atoms, which is critical to achieving the optimal interaction with the IN catalytic site. All of the newly designed compounds were characterized by introducing different substituents in various positions of phenyl rings. To better understand how these compounds block HIV replication, we docked the designed compounds into the HIV integrase protein.
 and our designed structures (8a-g, 9a-c, and 10a-f)")
Figure 2.
Structure of lead compounds (1, 2, and 3) and our designed structures (8a-g, 9a-c, and 10a-f)
2. Methods
2.1. General
All chemicals and solvents utilized in this study were of the highest quality and obtained from Sigma-Aldrich (Merck KGaA). An Electrothermal-IA9300 melting point device measured the compounds' melting points. A Perkin-Elmer infrared spectrometer (Model 1420) recorded the infrared spectra. The 1H and 13C NMR spectra were recorded using tetramethylsilane (TMS) as the internal standard in a Bruker FT-500 MHz NMR spectrometer (500 MHz for 1H; 126 MHz for 13C). DMSO-d6 was the solvent in this operation. Chemical shifts (d) were measured in parts per million (ppm), and the coupling constant (J) was expressed in hertz (Hz). A 6410 Agilent LCMS device with electrospray ionization (ESI) interface acquired high-resolution mass spectra. A Costech Elemental Combustion System 4010 elemental analyzer analyzed the elements C, H, and N. Microanalysis findings were within 0.4% of theoretical values.
2.2. Synthesis of diethyl 2-((o-tolylamino)methylene)malonate (5)
In the first step, a mixture of ortho-toluidine (4) (3 mL, 27.9 mmol) and ethoxy Methylene Malonate Diethyl Ester (EMME) (5.6 mL, 28 mmol) was prepared and heated for roughly two hours at 120°C. After completion of the reaction, the mixture was gradually cooled, and the precipitate was washed with n-hexane. The obtained product entered the next step after purification.
Yield: 75%; white powder; mp: 61.7 - 62.8°C; IR (KBr, spectroscopic grade): 1400 - 1600 (aromatic), 1629, 1670 (C=O), 3273 (N-H) cm-1; LC-MS (ESI): m/z 300.00 [M+Na]+, 577.10 [2M+Na]+
2.3. Synthesis of ethyl 8-methyl-4-oxo-4,1-dihydroquinoline-3-carboxylate (6)
Compound 5 was dissolved in diphenyl ether, and 2-chlorobenzoic acid was used as the catalyst. Next, the reaction solution was microwave irradiated for two hours at > 250°C. TLC was used to control the product formation. After the reaction was completed, the resulting mixture was cooled gradually until a precipitate formed, which was then filtered and washed with a large amount of n-hexane. The separated solid mass was recrystallized from ethanol.
Yield: 70%; white powder; mp: 266.5 - 267.8ºC; IR (KBr, spectroscopic grade): 1400 - 1600 (C=C aromatic), 1700 (C=O) cm-l; LCMS (ESI): m/z 229.8 [M-H]-.
2.4. Synthesis of 8-methyl-4-oxo-1,4-dihydroquinoline-3-carbohydrazide (7)
Compound 6 (2 g, 8.6 mmol) was dissolved in dimethylformamide (DMF) (8 mL), and hydrazine hydrate (4.2 mL, 86.49 mmol) was progressively added. The mixture was stirred at room temperature for 24 hours. After the reaction was completed, the mixture was poured into a container with water and ice and stored at room temperature for four hours. The obtained precipitate was then filtered and washed with a large amount of water. The solid mass was recrystallized from ethanol.
Yield: 80%; white powder; mp: 252°C; IR (KBr, spectroscopic grade): 1400 - 1600 (C=C aromatic), 1670 (C=O), 3300 (NH) cm-l; LCMS (ESI): m/z 215.8 [M-H]-.
2.5. General Procedure for the Synthesis of N'-[benzoyl] N'-8-methyl-4-oxo-1,4-dihydroquinoline-3-carbohydrazide Series (8a-g)
At ambient temperature, a mixture of compound 7 (0.30 g, 1.38 mmol), substituted benzoyl chloride (1.38 mmol), and Na2CO3 (0.4 mmol) was prepared in DMF and stirred for 17 hours. After completion of the reaction, the mixture was slowly poured over crushed ice. The solid was filtered, washed with water, and then recrystallized from ethanol to obtain the final compound.
2.5.1. N'-benzoyl- 8-methyl- 4-oxo- 1,4-dihydroquinoline- 3-carbohydrazide (8a)
Yield: 50%; white powder; mp: 274 - 275ºC; IR (KBr, spectroscopic grade): 1400 - 1600 (aromatic), 1630 (carbonyl), 3276 (N-H) cm-1; LCMS (ESI): m/z 322.10 [M+H+], 344.10 [M+Na+]; 1H NMR (500 MHz, DMSO-d6): δ ppm 2.53 (s, 3H, CH3), 7.36-7.39 (t, 1H, benzoyl H4' J = 7.7 Hz), 7.46-7.49 (t, 2H, benzoyl H3' & H5' , J = 7.4 Hz), 7.54-7.56 (t, 1H, H6, J = 7.3 Hz), 7.61 - 7.63 (d, 1H, H7, J = 7.0 Hz), 7.88 - 7.90 (d, 2H, benzoyl H2' & H6' J = 7.3 Hz), 8.14 - 8.15 (d, 1H, H5, J = 7.9 Hz), 8.61 (s, 1H, H2), 10.76 (s, 1H, NH), 11.69 (s, 1H, NH), 12.11 (br s, 1H, enolic OH). 13C NMR (DMSO-d6, 126 MHz): δ 17.53, 110.32, 123.88, 125.35, 126.64, 127.90, 128.02, 128.93, 132.28, 132.88, 134.20, 138.12, 143.91, 164.01, 165.29, 176.50; Anal. Calcd. for C18H15N3O3: C, 67.28; H, 4.71; N, 13.08. Found: C, 67.37; H, 4.91; N, 13.22.
2.5.2. N'-(4-fluorobenzoyl)- 8-methyl- 4-oxo- 1,4-dihydroquinoline- 3-carbohydrazide (8b)
Yield: 44.9%; mp: 291 - 293ºC; IR (KBr, spectroscopic grade): 1400 - 1600 (aromatic), 1600,1623 (carbonyl), 3247 (N-H) cm-1; MS (ESI): m/z 338 [M-H]-; 1H NMR (500 MHz, DMSO-d6): δ ppm 2.58 (s, 3H, CH3), 7.35 - 7.38 (t, 2H, 4-fluorobenzoyl H3' & H5', J = 8.8 Hz), 7.40 - 7.42 (t, 1H, H6, J = 7.8 Hz), 7.65 - 7.67 (d, 1H, H7, J = 7.1 Hz), 7.94 - 8.02 (dd, 2H, 4-fluorobenzoyl H2' & H6'), 8.17-8.18 (d, 1H, H5, J = 8.0 Hz), 8.65 (s, 1H, H2), 10.86 (s, 1H, NH), 11.75 (s, 1H, NH), 12.15 (br s, 1H, enolic OH). 13C NMR (DMSO-d6, 126 MHz): δ 17.52, 110.26, 115.95, 123.87, 125.35, 126.62, 127.91, 129.36, 130.74, 134.20, 138.12, 143.92, 163.41, 164.12, 165.89, 176.49. Anal. Calcd. for C18H14FN3O3: C, 63.71; H, 4.16; N, 12.38. Found: C, 63.56; H, 4.27; N, 12.64.
2.5.3. N'-(3-fluorobenzoyl)- 8-methyl- 4-oxo- 1,4-dihydroquinoline- 3-carbohydrazide (8c)
Yield: 75%; white powder; mp: 274.4 - 276.9°C; IR (KBr, spectroscopic grade): 1400 - 1600 (aromatic), 1620,1663 (carbonyl), 3280 (N-H) cm-1; LCMS (ESI): m/z 337.7 [M-H]-; 1H NMR (500 MHz, DMSO-d6): δ ppm 2.50 (s, 3H, CH3), 7.31 - 7.37 (dd, 1H, 3-fluorobenzoyl H4', J = 7.6 Hz), 7.39 - 7.41 (t, 1H, H6), 7.55-7.57 (m, 1H, , 3-fluorobenzoyl H5'), 7.66 - 7.68 (d, 1H, , 3-fluorobenzoyl H2', J = 9.5 Hz), 7.73 - 7.75 (d, 1H, H7, J = 7.8 Hz), 8.06 - 8.08 (d, 1H, 3-fluorobenzoyl H6', J = 7.9 Hz), 8.12 - 8.13 (d, 1H, H5, J = 7.9 Hz), 8.60 (s, 1H, H2), 10.64 (s, 1H, NH), 11.72 (s, 1H, NH), 12.2 (br s, 1H, enolic OH). 13C NMR (DMSO-d6, 126 MHz): δ 17.52, 110.20, 114.82, 119.24, 123.86, 124.23, 125.37, 126.62, 127.93, 131.20, 134.21, 135.15, 138.13, 143.95, 161.16, 163.59, 165.96, 176.48. Anal. Calcd. for C18H14FN3O3: C, 63.71; H, 4.16; N, 12.38. Found: C, 63.49; H, 4.25; N, 12.54.
2.5.4. N'-(4-chlorobenzoyl)- 8-methyl- 4-oxo- 1,4-dihydroquinoline- 3-carbohydrazide (8d)
Yield: 39%; white powder; mp: 304 - 305.2ºC; IR (KBr, spectroscopic grade): 1400 - 1600 (aromatic), 1637,1665 (carbonyl), 3295 (N-H) cm-1; LCMS (ESI): m/z 354 [M-H]-; 1H NMR (500 MHz, DMSO-d6): δ ppm 2.58 (s, 3H, CH3), 7.42-7.45 (t, 1H, H6, J =7.5 Hz), 7.60 - 7.62 (d, 2H, 4-chlorobenzoyl H3' & H5', J = 8.5 Hz), 7.67 - 7.68 (d, 1H, H7, J = 7.0 Hz), 7.94 - 7.96 (d, 2H, 4-chlorobenzoyl H2' & H6', J = 8.5 Hz), 8.18 - 8.20 (d, 1H, H5, J = 7.9 Hz), 8.65 (s, 1H, H2), 10.93 (s, 1H, NH), 11.77 (s,1H, NH), 11.80 (br s, 1H, enolic OH). 13C NMR (DMSO-d6, 126 MHz): δ 17.52, 110.24, 123.87, 125.33, 126.61, 127.88, 129.04, 129.95, 131.61, 134.19, 137.12, 138.11, 143.91, 163.93, 164.24, 176.48. Anal. Calcd. for C18H14ClN3O3: C, 60.77; H, 3.97; N, 11.81. Found: C, 60.95; H, 3.75; N, 11.61.
2.5.5. 8-Methyl-N'-(4-methylbenzoyl)- 4-oxo- 1,4-dihydroquinoline- 3-carbohydrazide (8e)
Yield: 80%; white powder; mp: 260 - 261.4°C; IR (KBr, spectroscopic grade): 1400 - 1600 (aromatic),1630,1673 (carbonyl), 3298 (N-H) cm-1; LCMS (ESI): m/z 336.10 [M+H+]; 1H NMR (500 MHz, DMSO-d6): δ ppm 2.39 (s, 3H, CH3), 2.58 (s, 3H, CH3), 7.32 - 7.33 (d, 2H, 4-methylbenzoyl H3' & H5', J = 7.9 Hz), 7.41 - 7.44 (t, 1H, H6, J = 7.6 Hz), 7.66 - 7.68 (d, 1H, H7, J = 7.1 Hz ), 7.83 - 7.84 (d, 2H, 4-methylbenzoyl H2' & H6', J = 7.9 Hz), 8.17 - 8.19 (d, 1H, H5, J = 8.0 Hz), 8.65 (s, 1H, H2), 10.72 (s, 1H, NH), 11.70 (s, 1H, NH), 12.15 (br s, 1H, enolic OH). 13C NMR (DMSO-d6, 126 MHz): δ 17.52, 21.50, 110.33, 123.86, 125.31, 126.62, 127.89, 128.03, 129.44, 130.06, 134.16, 138.11, 142.26, 143.88, 164.01, 165.19, 176.48. Anal. Calcd. for C19H17N3O3: C, 68.05; H, 5.11; N, 12.53. Found: C, 68.25; H, 5.26; N, 12.72.
2.5.6. 8-Methyl-N'-(3-methylbenzoyl)- 4-oxo- 1,4-dihydroquinoline- 3-carbohydrazide (8f)
Yield: 50%; white powder; mp: 292 - 293.7ºC; IR (KBr, spectroscopic grade): 1400 - 1600 (aromatic), 1650,1670 (carbonyl), 3277,3396 (N-H) cm-1; LCMS (ESI): m/z 334 [M-H]-; 1H NMR (500 MHz, DMSO-d6): δ ppm 2.34 (s, 3H, CH3), 2.53 (s, 3H, CH3), 7.34 - 7.40 (m, 3H, H6, 3-methylbenzoyl H4' & H5'), 7.61 - 7.63 (d, 1H, H7, J = 7.1 Hz), 7.67 - 7.70 (m, 2H, 3-methylbenzoyl H2' & H6'), 8.13 - 8.15 (d, 1H, H5, J = 8.0 Hz), 8.60 (s, 1H, H2), 10.69 (s, 1H, NH), 11.68 (s, 1H, NH), 12.12 (br s, 1H, enolic OH). 13C NMR (DMSO-d6, 126 MHz): δ 17.53, 21.41, 110.33, 123.89, 125.12, 125.38, 126.65, 127.92, 128.57, 128.81, 132.86, 134.22, 138.14, 138.22, 143.92, 163.93, 165.37, 176.51. Anal. Calcd. for C19H17N3O3: C, 68.05; H, 5.11; N, 12.53. Found: C, 68.25; H, 5.29; N, 12.74.
2.5.7. N'-(4-methoxybenzoyl)- 8-methyl- 4-oxo- 1,4-dihydroquinoline- 3-carbohydrazide (8g)
Yield: 70%; white powder; mp: 274ºC (decomposed); IR (KBr, spectroscopic grade): 1400 - 1600 (aromatic), 1678 (carbonyl), 3276 (N-H) cm-1; LCMS (ESI): m/z 349.70 [M-H]-; 1H NMR (500 MHz, DMSO-d6): δ ppm 2.59 (s, 3H, CH3), 3.84 (s, 3H, OCH3), 7.05 - 7.06 (d, 2H, 4-methoxybenzoyl H3' & H5', J = 8.8 Hz), 7.42 - 7.45 (t, 1H, H6, J = 7.5 Hz), 7.67 - 7.68 (d, 1H, H7, J = 7.1 Hz), 7.91 - 7.93 (d, 2H, 4-methoxybenzoyl H2' & H6', J = 8.7 Hz), 8.18 - 8.20 (d, 1H, H5, J = 7.9 Hz), 8.65 (s, 1H, H2), 10.65 (s, 1H, NH), 11.68 (s, 1H, NH), 12.16 (br s, 1H, enolic OH). 13C NMR (DMSO-d6, 126 MHz): δ 17.52, 55.85, 110.38, 114.14, 123.88, 124.98, 125.33, 126.64, 127.89, 129.90, 134.18, 138.12, 143.88, 162.45, 164.04, 164.81, 176.50. Anal. Calcd. for C19H17N3O4: C, 64.95; H, 4.88; N, 11.96. Found: C, 64.75; H, 5.02; N, 11.81.
2.6. General Procedure for the Synthesis of N-(phenyl)- 2-(8-methyl- 4-oxo- 1,4-dihydroquinoline- 3-carbonyl) hydrazine- 1-carboxamide Series (9a-c)
Compound 7 (0.22 g, 0.1 mmol) was dissolved in dry DMF (4 mL). Then, isocyanate derivatives (0.1 mmol) were progressively added to the mixture. The reaction solution was stirred for 24 hours at room temperature. Thin-layer chromatography was used to monitor and control the product. Water and ice were poured into the reaction to form a precipitate, which was then filtered through a Buchner funnel and thoroughly rinsed with plenty of water. The precipitate was crystallized using 96% ethanol solution.
2.6.1. 2-(8-Methyl- 4-oxo- 1,4-dihydroquinoline- 3-carbonyl)- N-phenylhydrazine- 1-carboxamide (9a)
Yield: 73%; white powder; mp: 310.9°C (decomposed); IR (KBr, spectroscopic grade): 1400 - 1600 (aromatic), 1641,1676 (C=O), 2589 - 3500 (OH), 3338, 3462 (NH) cm-1; LCMS (ESI): m/z 337.0 [M+H+]; 1H NMR (500 MHz, DMSO-d6): δ ppm 2.57 (s, 3H, CH3), 6.94 - 6.98 (t, 1H, N-phenyl H4' , J = 8.0 Hz), 7.24 - 7.28 (t, 2H, N-phenyl H3' & H5', J = 8.0 Hz), 7.40 - 7.44 (t, 1H, H6, J = 8.0 Hz), 7.47 (d, 2H, N-phenyl H2' & H6', J = 8.0 Hz) 7.65 (d, 1H, H7, J = 4.0 Hz), 8.17 (d, 1H, H5, J = 8.0 Hz), 8.45 (br s, 1H, NH), 8.61 (s, 1H, H2), 8.92 (s, 1H, NH), 11.41 (br s, 1H, NH), 12.14 (br s, 1H, enolic OH); 13C NMR (DMSO-d6, 126 MHz): δ 17.51, 110.45, 118.79, 122.32, 123.88, 125.30, 126.69, 127.86, 129.14, 134.16, 138.12, 140.12, 143.78, 155.34, 164.45, 176.44; Anal. Calcd. for C18H16N4O3: C, 64.28; H, 4.79; N, 16.66. Found: C, 64.42; H, 4.66; N, 16.85.
2.6.2. N-benzyl- 2-(8-methyl- 4-oxo- 1,4-dihydroquinoline- 3-carbonyl) hydrazine- 1-carboxamide (9b)
Yield: 70%; white powder; mp: 280.1°C (decomposed); IR (KBr, spectroscopic grade): 1400 - 1600 (aromatic), 1653 (C=O), 2783 - 3429 (OH), 3310 (NH) cm-1; LCMS (ESI): m/z 351 [M+H+] ,372.9 [M+Na+]; 1H NMR (500 MHz, DMSO-d6): δ ppm 2.56 (s, 3H, CH3), 4.25 (d, 2H, -CH2, J = 4.0 Hz), 7.07 (br s, 1H, NH), 7.21 - 7.24 (t, 1H, N-benzyl H4', J = 8.0 Hz), 7.27 - 7.34 (m, 4H, N-benzyl H2', H3', H5' & H6'), 7.38-7.42 (t, 1H, H6, J = 8.0 Hz),7.65 (d, 1H, H7, J = 8.0 Hz), 8.15 (d, 1H, H5, J = 8.0 Hz), 8.22 (br s, 1H, NH), 8.61 (s, 1H, H2), 11.26 (br s, 1H, NH), 12.12 (br s,1H, enolic OH); 13C NMR (DMSO-d6, 126 MHz): δ 17.51, 110.65, 123.88, 125.25, 126.69, 127.02, 127.44, 127.81, 128.62, 134.12, 138.09, 140.99, 143.73, 158.38, 164.72, 176.43; Anal. Calcd. for C19H18N4O3: C, 65.13; H, 5.18; N, 15.99. Found: C, 65.25; H, 5.33; N, 15.78.
2.6.3. N-(3,4-dichlorophenyl)- 2-(8-methyl- 4-oxo- 1,4-dihydroquinoline- 3-carbonyl) hydrazine- 1-carboxamide (9c)
Yield: 62%; white powder; mp: 292.4°C (decomposed); IR (KBr, spectroscopic grade): 1400 - 1600 (aromatic), 1648, 1686 (C=O), 2587 - 3500 (OH), 3335, 3488 (NH) cm-1; LCMS (ESI): m/z 405.0 [M+H+]; 1H NMR (500 MHz, DMSO-d6): δ ppm 2.57 (s, 3H, CH3), 7.40 - 7.44 (t, 2H, H6, J = 8.0 Hz), 7.49 - 7.50 (d, 1H, 3,4-dichlorophenyl H6', J = 8.0 Hz), 7.65 - 7.67 (d, 1H, H7, J = 8.0 Hz), 7.87 (d, 1H, 3,4-dichlorophenyl H5', J = 8.0 Hz), 8.17 (d, 1H, H5, J = 8.0 Hz), 8.61 (s, 1H, H2), 8.64 (s, 1H, 3,4-dichlorophenyl H2'), 9.24 (s,1H, NH), 11. 38 (br s, 1H, NH), 12.16 (br s, 1H, enolic OH); 13C NMR (DMSO-d6, 126 MHz): δ 17.52, 110.43, 118.97, 119.99, 123.57, 123.88, 125.36, 126.70, 127.89, 130.92, 131.31, 134.21, 138.12, 140.49, 143.86, 176.46; Anal. Calcd. for C18H14Cl2N4O3: C, 53.35; H, 3.48; N, 13.83. Found: C, 53.52; H, 3.59; N, 13.99.
2.7. General Procedure for the Synthesis of 2-(8-dimethyl- 4-oxo- 1,4-dihydroquinoline- 3-carbonyl)- N-phenylhydrazine- 1-carbothioamide (10a-f)
Compound 7 (0.22 g, 0.1 mmol) was dissolved in dry DMF (4 mL). Next, isothiocyanate derivatives (0.1 mmol) were progressively added to the mixture. The reaction solution was stirred for 4 hours at room temperature. Thin-layer chromatography was used to monitor and control the product. Water and ice were poured into the reaction to form a precipitate, which was then filtered through a Buchner funnel and thoroughly rinsed with plenty of water. The precipitate was crystallized using 96% ethanol solution.
2.7.1. 2-(8-Methyl- 4-oxo- 1,4-dihydroquinoline- 3-carbonyl)- N-phenylhydrazine- 1-carbothioamide (10a)
Yield: 80%; white powder; mp: 237.6°C (decomposed); IR (KBr, spectroscopic grade): 1400 - 1600 (aromatic), 1646,1662 (C=O), 2765 - 3392 (OH), 3224, 3324 (NH) cm-1; LCMS (ESI): m/z 374.8 [M+Na+], 353.0 [M+H]+; 1H NMR (500 MHz, DMSO-d6): δ ppm 2.58 (s, 3H,CH3), 7.12 - 7.16 (t, 1H, N-phenyl H4' , J = 8.0 Hz), 7.31 - 7.35 (t, 2H, N-phenyl H3' & H5', J = 8.0 Hz), 7.40 - 7.44 (t, 1H, H6, J = 8.0 Hz), 7.53 (m, 2H, N-phenyl H2' & H6'), 7.66 (d, 1H, H7, J = 8.0 Hz), 8.18 (d, 1H, H5, J = 8.0 Hz), 8.62 (s, 1H, H2), 9.87 (br s, 2H, NH), 12.16 (br s, 1H, enolic OH), 13C NMR (DMSO-d6, 126 MHz): δ 17.52, 123.89, 125.36, 126.64, 127.87, 128.72, 134.22, 138.10, 139.73, 143.71, 176.33; Anal. Calcd. for C18H16N4O2S: C, 61.35; H, 4.58; N, 15.90. Found: C, 61.42; H, 4.80; N, 15.70.
2.7.2. 2-(8-Methyl- 4-oxo- 1,4-dihydroquinoline- 3-carbonyl)- N-(o-tolyl) hydrazine- 1-carbothioamide (10b)
Yield: 81%; white powder; mp: 237.1°C (decomposed); IR (KBr, spectroscopic grade): 1400 - 1600 (aromatic), 1648, 1670 (C=O), 2878 - 3400 (OH), 3130, 3290 (NH) cm-1; LCMS (ESI): m/z 367.0 [M+H+]; 1H NMR (500 MHz, DMSO-d6): δ ppm 2.21 (s, 3H, CH3), 2.57 (s, 3H, 8-CH3), 7.13 - 7.22 (m, 4H, N-o-tolyl H3', H4', H5' & H6'),7.40 - 7.44 (t, 1H, H6, J = 8.0 Hz), 7.66 (d, 1H, H7, J = 8.0 Hz), 8.17 (d, 1H, H5, J = 8.0 Hz), 8.62 (s, 1H, H2), 9.53 (br s, 1H, NH), 9.72 (br s, 1H, NH), 11. 49 (br s, 1H, NH), 12.15 (br s, 1H, enolic OH), 13C NMR (DMSO-d6, 126 MHz): δ 17.53, 18.18, 123.89, 125.33, 126.64, 126.88, 127.85, 130.57, 134.20, 138.09, 143.67, 158.38, 164.72, 176.43; Anal. Calcd. for C19H18N4O2S: C, 62.28; H, 4.95; N, 15.29. Found: C, 62.36; H, 4.85; N, 15.39.
2.7.3. 2-(8-Methyl- 4-oxo- 1,4-dihydroquinoline- 3-carbonyl)-N-(m-tolyl) hydrazine- 1-carbothioamide (10c)
Yield: 75.6%; white powder; mp: 212.6°C; IR (KBr, spectroscopic grade): 1400 - 1600 (aromatic), 1668 (C=O), 2759 - 3500 (OH), 3230, 3330 (NH) cm-1; LCMS (ESI): m/z 367 [M+H+]; 1H NMR (500 MHz, DMSO-d6): δ ppm 2.29 (s, 3H, CH3), 2.58 (s, 3H, 8-CH3), 6.95 (d, 1H, m-tolyl H4', J= 8.0 Hz), 7.19 - 7.23 (t, 1H, m-tolyl H5', J = 8.0 Hz), 7.34 (m, 2H, m-tolyl H2' & H6'), 7.40 - 7.44 (t, 1H, H6, J = 8.0 Hz), 7.66 (d, 1H, H7, J = 8.0 Hz), 8.18 (d, 1H, H5, J = 8.0 Hz), 8.62 (s, 1H, H2), 9.80 (br s, 2H, NH), 12.16 (d, 1H, enolic OH), 13C NMR (DMSO-d6, 126 MHz): δ 17.53, 21.47, 123.89, 125.36, 126.64, 127.87, 134.22, 138.09, 139.59, 143.70, 176.32; Anal. Calcd. for C19H18N4O2S: C, 62.28; H, 4.95; N, 15.29. Found: C, 62.39; H, 5.12; N, 15.39.
2.7.4. 2-(8-Methyl- 4-oxo- 1,4-dihydroquinoline- 3-carbonyl)- N-(p-tolyl) hydrazine- 1-carbothioamide (10d)
Yield: 85.6%; white powder; mp: 232.4°C (decomposed); IR (KBr, spectroscopic grade): 1400 - 1600 (aromatic), 1615,1639 (C=O), 2757 - 3379 (OH), 3210, 3250 (NH) cm-1; LCMS (ESI): m/z 367 [M+H+]; 1H NMR (500 MHz, DMSO-d6): δ ppm 2.28 (s, 3H, CH3), 2.57 (s, 3H, 8-CH3), 7.13 (d, 2H, p-tolyl H3' & H5', J = 8.0 Hz), 7.38 - 7.44 (m, 3H, H6 and p-tolyl H2' & H6'),7.66 (d, 1H, H7, J = 8.0 Hz), 8.17 (d, 1H, H5, J = 8.0 Hz), 8.61 (s, 1H, H2), 9.79 (br s, 2H, NH), 12.15 (br s, 1H, enolic OH), 13C NMR (DMSO-d6, 126 MHz): δ 17.52, 21.00, 123.89, 125.35, 126.64, 127.86, 129.17, 134.21, 137.11, 138.09, 143.69, 176.33; Anal. Calcd. for C19H18N4O2S: C, 62.28; H, 4.95; N, 15.29. Found: C, 62.35; H, 4.73; N, 15.09.
2.7.5. 2-(8-Methyl- 4-oxo- 1,4-dihydroquinoline- 3-carbonyl)- N-(4-fluorophenyl) hydrazine- 1-carbothioamide (10e)
Yield: 81%; white powder; mp: 239.7°C (decomposed); IR (KBr, spectroscopic grade): 1400 - 1600 (aromatic), 1647, 1662 (C=O), 2789 - 3500 (OH), 3218 (NH) cm-1; LCMS (ESI): m/z 370.8 [M+H+]; 1H NMR (500 MHz, DMSO-d6): δ ppm 2.58 (s, 3H, CH3), 7.14 - 7.19 (t, 2H, 4-fluorophenyl H3' & H5', JH,F = 12.0 Hz), 7.40 - 7.44 (t, 1H, H6, J = 8.0 Hz), 7.49 (m, 2H, 4-fluorophenyl H2' & H6'), 7.66 (d, 1H, H7, J = 8.0 Hz), 8.17 (d, 1H, H5, J = 8.0 Hz), 8.62 (s, 1H, H2), 9.85 (br s, 2H, NH), 12.17 (br s, 1H, enolic OH), 13C NMR (DMSO-d6, 126 MHz): δ 17.53, 115.36, 123.89, 125.39, 126.64, 127.89, 134.24, 136.05, 138.10, 143.75, 176.35. Anal. Calcd. for C18H15FN4O2S: C, 58.37; H, 4.08; N, 15.13. Found: C, 58.55; H, 4.22; N, 15.28.
2.7.6. N-(4-methoxyphenyl)- 2-(8-methyl- 4-oxo- 1,4-dihydroquinoline- 3-carbonyl) hydrazine- 1-carbothioamide (10f)
Yield: 70.2%; white powder; mp: 230.7 - 233.3°C (decomposed); IR (KBr, spectroscopic grade): 1400 - 1600 (aromatic), 1648 (C=O), 2795 - 3395 (OH), 3259 (NH) cm-1; LCMS (ESI): m/z 383.1 [M+H+]; 1H NMR (500 MHz, DMSO-d6): δ ppm 2.57 (s, 3H, CH3), 3.74 (s, 3H, OCH3), 6.90 (d, 2H, 4-methoxyphenyl H3' & H5', J = 8.0 Hz), 7.35 (d, 2H, 4-methoxyphenyl H2' & H6', J = 8.0 Hz), 7.40-7.44 (t, 1H, H6, J = 8.0 Hz), 7.66 (d, 1H, H7, J = 8.0 Hz), 8.17 (d, 1H, H5, J = 8.0 Hz), 8.61 (s, 1H, H2), 9.72 (br s, 2H, NH), 12.14 (br s, 1H, enolic OH); Anal. Calcd. for C19H18N4O3S: C, 59.67; H, 4.74; N, 14.65. Found: C, 59.53; H, 4.66; N, 14.38.
2.8. Molecular Docking Study
The docking study was performed using the high-resolution X-ray diffraction structure of the Prototype Foamy Virus (PFV) crystal (PDB code: 3OYA) in association with Mg+2 and raltegravir. To prepare the ligand and perform conformational analysis, we utilized HyperChem 8.0.10. Protein and ligands were prepared using Autodock tools 1.5.6 (ADT) from the molecular graphics laboratory (MGL) Tools (34). In this simulation, water molecules were removed to simplify calculations. The co-crystallized ligand was extracted, Kollman united partial charges were added, and non-polar hydrogens were merged. The active site was defined as a grid box with dimensions of 20 × 20 × 20 Å enclosing the crystallographic ligand raltegravir. The docking used in this study is a semi-flexible method of operation. AutoDock4 software was used to insert ligands in the enzyme's binding site.
2.9. In Vitro Anti-HIV-1 and Cytotoxicity Assay Method
The synthesized compounds were evaluated for anti-HIV activity by single-cycle replication assay as reported previously. As part of this study, an HIV-1 genome-mutated in the poly (pol) gene was co-transfected with plasmids that expressed reverse transcriptase (RT) and integrase (IN), as well as glycoprotein G of the vesicular stomatitis virus (VSV). The virions produced in HEK 293 T cells were antigenic but only replicated for one cycle (first-generation single-cycle replicable (SCR) virions). DMSO was used to dissolve the compounds at concentrations ranging from 10 mmol/L to 10 μmol/L. These stocks were diluted 100 times in the cell environment, resulting in final compound concentrations ranging from 100 μmol/L to 10 nmol/L. We used zidovudine (AZT) and DMSO (1% v/v) as positive and negative controls in the experiment, respectively. All tests were run three times. HeLa cells (6 × 103 per well of 96-well plates) were infected with single-cycle replicable HIV NL4-3 virions (200 ng p24) in the presence of various concentrations of compounds. Compounds were added to the cell environment simultaneously with a viral infection. Capture ELISA (Biomerieux, France) measured the p24 antigen load in cell culture supernatant 72 hours after infection. The inhibition rate of p24 (%) was used to compute the percentage inhibition of p24 expression in the treated culture. The XTT proliferation technique was used to evaluate the cellular toxicity of substances. The XTT (sodium 3-[1 (phenyl aminocarbonyl)-3,4-tetrazolium]-bis(4-methoxy-6-nitro) benzene sulfonic acid) reagent was used according to the kit instruction (Roche, Germany). In a CO2 incubator, cells were cultured in 96-well plates (3,5104 cells/well) with fresh phenol-red free medium and incubated for 72 hours. Next, 50 μL of XTT was added to each well and incubated for four hours at 37°C. Plates were evaluated by an ELISA reader at the test and reference OD of 450 and 630 nm, respectively. The cytotoxic concentration that reduced the number of viable cells by 50% (CC50) was calculated after determining the p24 load in HIV-1 replication assay plates (35-38).
3. Results and Discussion
3.1. Chemistry
The synthesis of the target compounds is described in Figure 3. O-toluidine (4) was used to begin the synthesis of the target compounds. Compound 5 (diethyl 2-((o-tolylamino) malonate) was synthesized by condensing o-toluidine with EMME. Compound 5 was turned into ethyl 8-methyl-4-oxo-1,4-dihydroquinoline-3-carboxylate (6) in diphenyl ether containing catalytic 2-chlorobenzoic acid. A DMF solution of hydrazine was used to make 8-methyl-4-oxo-1,4-dihydroquinoline-3-carbohydrazide (compound 7) from compound 6. Compound 7 was the primary intermediate for synthesizing three series of substances. Compound 7 interacted with substituted benzoyl chlorides in DMF with a catalytic quantity of Na2CO3 to produce N'-[substituted benzoyl] N'-8-methyl-4-oxo-1,4-dihydroquinoline-3-carbohydrazide derivatives (8a-g). The second and third series of compounds were prepared by the reaction of compound 7 with the phenyl isocyanate and phenyl isothiocyanate derivatives in dry DMF to produce compounds 9a-c and 10a-f, respectively. The structure of synthesized compounds was determined by 1H-NMR, 13C-NMR, LC-MS technique, and elemental analysis.
 EMME, 125°C, 90 min; (ii) 2-chlorobenzoic acid, Ph<sub>2</sub>O, 250°C, 120 min; (iii) NH<sub>2</sub>NH<sub>2</sub>, DMF, rt; (iv) benzoyl chloride derivatives, Na<sub>2</sub>CO<sub>3</sub>, DMF, rt, 17 h; (v) phenyl isocyanate derivatives, dry DMF, rt, 4 h; (vi) phenyl isothiocyanate derivatives, dry DMF, rt, 24 h")
Figure 3.
Reagents and conditions: (i) EMME, 125°C, 90 min; (ii) 2-chlorobenzoic acid, Ph2O, 250°C, 120 min; (iii) NH2NH2, DMF, rt; (iv) benzoyl chloride derivatives, Na2CO3, DMF, rt, 17 h; (v) phenyl isocyanate derivatives, dry DMF, rt, 4 h; (vi) phenyl isothiocyanate derivatives, dry DMF, rt, 24 h
3.2. Biological Results
The novel 8-methyl-4-oxo-1,4-dihydroquinoline-3-carbohydrazide derivatives featuring various substituted benzoyl and N-phenyl carboxamide or carbothioamide moieties were synthesized and evaluated for their potential to prevent single-cycle replicable (SCR) HIV-1 (NL4-3) replication in HeLa cell culture. In addition, the cytotoxicity of the compounds was controlled to ensure that the anti-HIV activity was entirely free of cytotoxic activity. Zidovudine (AZT) was used as a positive control. Figure 4 summarizes the biological data of the synthesized compounds reported as EC50 and CC50.
All tested compounds exhibited CC50 > 500 µM, suggesting no significant cytotoxicity and a good safety profile according to the biological results. The antiviral activity profile of the compounds demonstrated that the synthesized compounds displayed antiviral activity with EC50 values ranging from 75 to 150 µM, which were lower than the reference drug. Compounds 8a-g possessing substituted benzoyl moieties were more active than compounds 9a-c and 10a-f containing N-phenyl carboxamide or carbothioamide groups. It can be concluded that the elongation of the linker decreased the anti-HIV activity. As shown in Figure 4, the variation of the substituents on the phenyl ring of compounds 8a-g, 9a-c, and 10a-f influenced the anti-HIV activity in the following order: F > Me > OMe > Cl > H. In addition, in compounds 8a-g, the position of substituents on the benzoyl ring affected the potency of compounds, as flour and methyl groups at the para-position showed slightly better activity than the meta-position. This agrees with the SAR of HIV IN inhibitors that generally contain flour groups on the hydrophobic portion. The best anti-HIV activity was observed for compound 8b containing 4-flourobenzoyl moiety with an EC50 of 75 µM.

Figure 4.
Anti-HIV activity, cytotoxicity, and binding energy of compounds 8a-g, 9a-c, and 10a-f
3.3. Molecular Docking Study
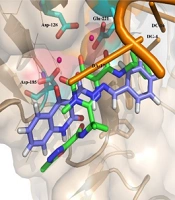
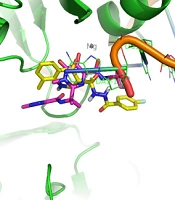
Docking tests were carried out on the integrase active site to determine how the compounds under study may interact. The binding mode of substances in the IN active site was studied using compound 8b. AutoDock4 was used for the docking study, while PyMOL software was used to visualize and analyze the results. For docking studies, we utilized the PFV-IN structure in a complex with two magnesium ions (Mg2+), raltegravir, and a double-stranded DNA (DS) (PDB code: 3OYA). It has been previously reported that the PFV-integrase and HIV integrase secondary structures have a high degree of homology with an RMSD of 1.04 (39). The co-crystallized ligand, raltegravir, was redocked to verify the method. To calculate RMSD, we used the PyMOL program to superimpose the optimal pose on the co-crystallized raltegravir. As shown in Figure 4, the designed compounds exhibited good binding energy ranging from -6.93 to -8.15 kcal/mol. Compound 8b fitted well in the active site, indicating possible interactions with Mg2+ via its carbonyl moieties (Figure 5). In the hydrophobic pocket, the benzoyl ring interacted with the 3'-end-DA17 through the π–π interactions. The flour group in the para position also could form hydrogen bonds at the active site (Figure 5). Overall, ligand 8b closely mimicked raltegravir's binding mode, as seen by a 3-D superposition of the two substances (Figure 6). According to docking studies, our molecules included the significant integrase pharmacophores inhibitors and may act via HIV integrase inhibition.
 (PDB 3OYA)")
Figure 5.
Compound 8b binding mode at the PFV IN active site (shown in yellow) (PDB 3OYA)
 is superimposed over co-crystallized raltegravir (shown in purple) in the PFV IN active site")
Figure 6.
Compound 8b (shown in yellow) is superimposed over co-crystallized raltegravir (shown in purple) in the PFV IN active site
4. Conclusions
The research focused on the design and synthesis of 8-methyl-4-oxo-1,4-dihydroquinoline-3-carbohydrazide derivatives characterized by various substituted benzoyl and N-phenyl carboxamide or carbothioamide moieties based on the essential pharmacophores of HIV IN inhibitors. The in vitro anti-HIV and cytotoxicity assays of the proposed compounds exhibited that they were active against HIV replication with EC50 values ranging from 75 to 150 µM and CC50 > 500 µM. The docking analysis demonstrated that the suggested compounds could interact with two Mg2+ ions and other critical residues in the active site of IN. Based on the low cytotoxicity and good anti-HIV activity, the 8-methyl-4-oxo-1,4-dihydroquinoline-3-carbohydrazide scaffold can be used for further development.