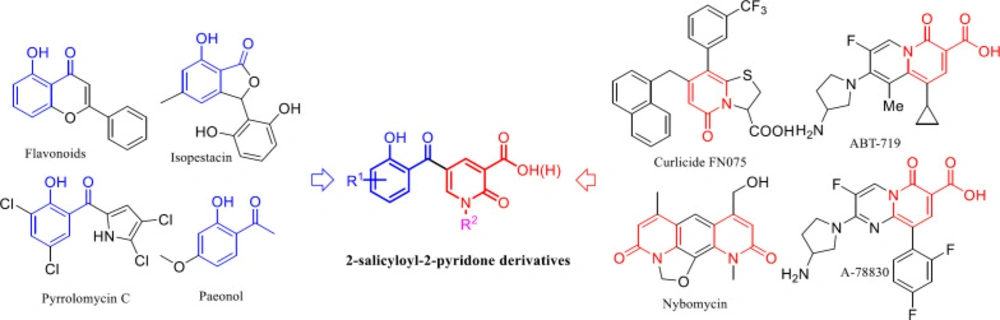
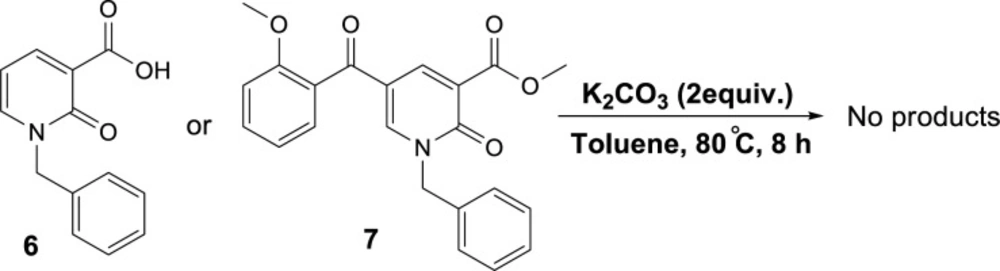
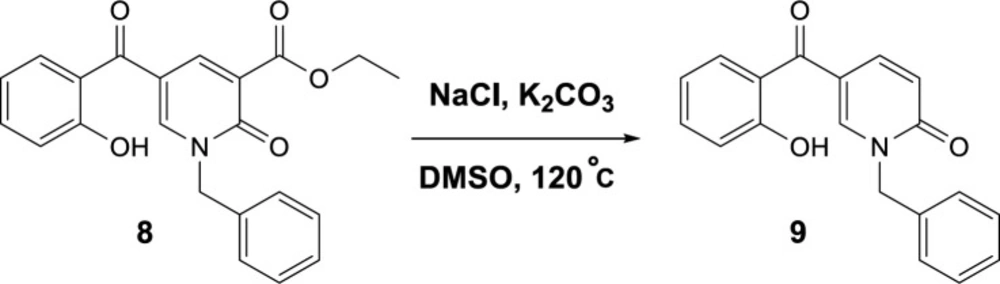
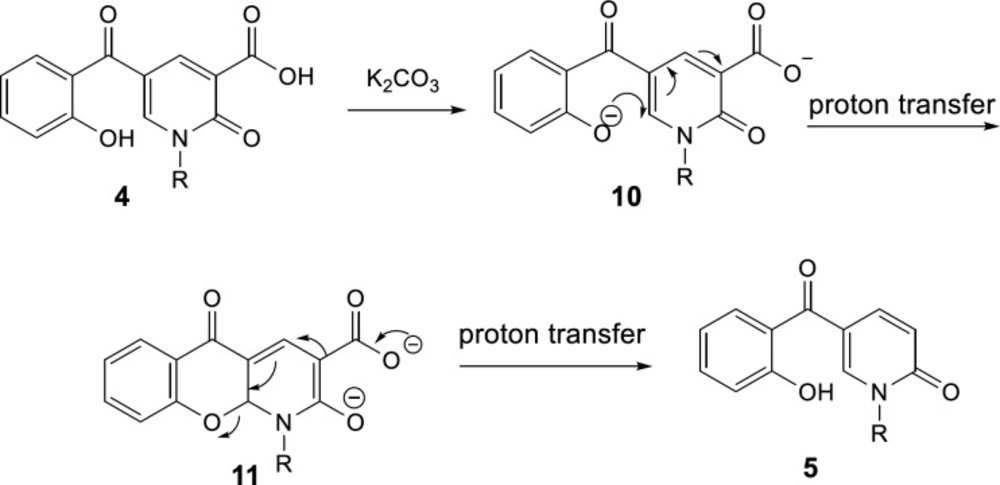
General
All organic reactions were done under the air atmosphere. Commercially available substances were procured in reagent grades with high purity. Thin-layer chromatography (TLC) was aluminum plates coated with silica gel 60-F254, and the reaction progress was monitored by it under UV light of 254 nm for detection. Flash column chromatography was filled with silica gel 63-200 mesh and employed to purify products 5a-o. The melting point was presented by the Electrothermal 9100 apparatus. 1H and 13C NMR spectroscopy were documented on a Bruker 600, 400, 300 MHz and 150, 100, 75 MHz, respectively. Chemical shifts (δ) of 1H and 13C NMR spectra were prepared as ppm, and coupling constants (J) have been reported in Hertz (Hz). High-resolution mass (ESI-HRMS) was obtained by Agilent Q-TOF LC-MS spectrometer.
General process of the 4a-p synthesis
Products
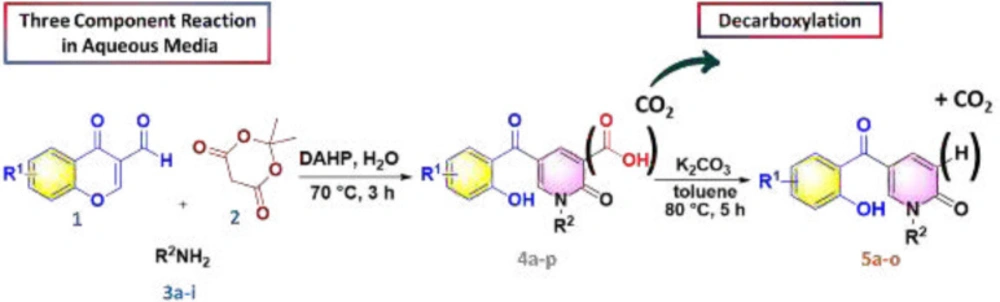
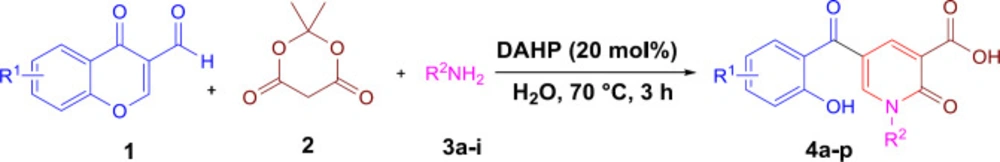
4a-p were prepared by the reported procedure (
25).
1-benzyl-5-(2-hydroxybenzoyl)-2-oxo-1,2-dihydropyridine-3-carboxylic acid (4a)
Yellow solid, mp 199-200 ℃, yield 86%; 1H NMR (400 MHz, DMSO-d6): δH = 13.51 (s, 1H, COOH), 10.43 (s, 1H, OH), 8.93 (d, J = 2.6 Hz, 1H, H-6-py), 8.51 (d, J = 2.5 Hz, 1H, H-4-py), 7.46 (t, J = 7.7 Hz, 1H, H-Ar), 7.41 – 7.31 (m, 6H, H-Ar), 7.02 (d, J = 8.2 Hz, 1H, H-Ar), 6.97 (t, J = 7.7 Hz, 1H, H-Ar), 5.40 (s, 2H, CH2N); 13C NMR (100 MHz, DMSO-d6): δC = 191.4, 164.9, 163.0, 156.3, 149.1, 144.8, 135.9, 133.9, 130.7, 129.2, 128.5, 128.4, 124.8, 120.0, 118.8, 117.2, 117.1, 53.6; LCMS-ESI (m/z) Calcd. for C20H16NO5 [M+H]+: 350.3500; found, 350.3.
5-(2-hydroxybenzoyl)-2-oxo-1-(prop-2-yn-1-yl)-1,2-dihydropyridine-3-carboxylic acid (4e)
Yellow solid, mp 135-136 ℃, yield 78%; 1H NMR (600 MHz, CDCl3): δH = 13.34 (s, 1H, COOH), 11.33 (s, 1H, OH), 8.88 (d, J = 2.0 Hz, 1H, H-6-py), 8.65 (d, J = 2.0 Hz, 1H, H-4-py), 7.56 (t, J = 7.8 Hz, 1H, H-Ar), 7.49 (d, J = 7.8 Hz, 1H, H-Ar), 7.10 (d, J = 8.4 Hz, 1H, H-Ar), 6.95 (t, J = 7.8 Hz, 1H, H-Ar), 4.95 (d, 2H, J = 2.6 Hz, CH2N), 2.73 (t, J = 2.7 Hz, 1H, HC≡C); 13C NMR (151 MHz, CDCl3): δC = 193.8, 163.6, 163.2, 162.9, 145.6, 144.3, 137.3, 131.5, 119.5, 119.4, 119.1, 118.1, 117.0, 74.1, 79.1, 39.8; HRMS-ESI (m/z) Calcd. for C16H12NO5 [M+H]+: 298.0610; found, 298.0630.
5-(2-hydroxybenzoyl)-2-oxo-1-((1-phenyl-1H-1,2,3-triazol-5-yl)methyl)-1,2dihydropyridine-carboxylic acid (4g)
White solid, mp 186-187 ℃, yield 69%; 1H NMR (300 MHz, CDCl3): δH = 13.53 (s, 1H, COOH), 11.31 (s, 1H, OH), 8.85 (d, J = 2.2 Hz, 1H, H-6-py), 8.68 (d, J = 2.1 Hz, 1H, H-4-py), 8.28 (s, 1H, CH-triazol), 7.73 (d, J = 7.8 Hz, 2H, H-Ar), 7.45 – 7.59 (m, 5H, H-Ar), 7.08 (d, J = 8.2 Hz, 1H, H-Ar), 6.96 (t, J = 7.6 Hz, 1H, H-Ar), 5.52 (s, 2H, CH2N); 13C NMR (75 MHz, CDCl3): δC = 193.8, 163.9, 163.7, 162.8, 146.4, 145.7, 140.9, 137.2, 136.6, 131.7, 129.9, 129.4, 122.6, 120.7, 119.6, 119.5, 118.9, 119.0, 117.4, 45.9; HRMS-ESI (m/z) Calcd. for C22H16N4NaO5 [M+Na]+: 439.1018 ; found, 439.1013.
1-(3,4-dimethoxyphenethyl)-5-(2-hydroxybenzoyl)-2-oxo-1,2-dihydropyridine-3-carboxylic acid (4h)
Yellow solid, mp 109-111 ℃, yield 74%; 1H NMR (300 MHz, CDCl3): 13.72 (s, 1H, COOH), 11.23 (s, 1H, OH), 8.83 (d, J = 2.5 Hz, 1H, H-6-py), 7.68 (d, J = 2.5 Hz, 1H, H-4-py), 7.46-7.53 (m, 1H, H-Ar), 7.02 (d, J = 8.3 Hz, 1H, H-Ar), 6.87 (dd, J = 8.1, 1.8 Hz, 1H, H-Ar), 6.83 (d, J = 7.7 Hz, 1H, H-Ar), 6.77 – 6.80 (m, 1H, H-Ar), 6.70 (d, J = 1.8 Hz, 1H, H-Ar), 6.58 (dd, J = 8.1, 1.8 Hz, 1H, H-Ar), 4.36 (t, J = 6.5 Hz, 2H, CH2N), 3.84 (s, 3H, OMe), 3.82 (s, 3H, OMe), 3.13 (t, J = 6.5 Hz, 2H, CH2Ph); 13C NMR (75 MHz, CDCl3): δC = 193.6, 164.1, 163.8, 162.7, 149.7, 148.5, 146.3, 145.4, 137.0, 131.2, 128.6, 121.3, 119.3, 118.9, 118.3, 118.0, 117.4, 111.8, 111.7, 56.0, 55.9, 54.1, 33.9; ; HRMS-ESI (m/z) Calcd. for C23H22NO7 [M+H]+ :424.1397; found, 424.1391.
5-(2-hydroxybenzoyl)-2-oxo-1-phenyl-1,2-dihydropyridine-3-carboxylic acid (4i)
Yellow solid, mp 207-209 ℃, yield 65%; 1H NMR (600 MHz, CDCl3): δH = 13.44 (s, 1H, COOH), 11.31 (s, 1H, OH), 8.95 (d, J = 2.2 Hz, 1H, H-6-py), 8.28 (d, J = 2.1 Hz, 1H, H-4-py), 7.52 – 7.63 (m, 5H, H-Ar), 7.44 (d, J = 7.4 Hz, 2H, H-Ar), 7.09 (d, J = 8.7 Hz, 1H, H-Ar), 6.96 (t, J = 7.6 Hz, 1H, H-Ar); 13C NMR (151 MHz, CDCl3): δC = 193.9, 163.9, 163.8, 162.9, 146.3, 145.9, 138.4, 137.3, 131.5, 130.5, 130.0, 126.1, 119.6, 119.4, 119.1, 118.1, 117.8; HRMS-ESI (m/z) Calcd. for C19H14NO5 [M+H]+: 336.0860; found, 336.0868.
1-benzyl-5-(5-fluoro-2-hydroxybenzoyl)-2-oxo-1,2-dihydropyridine-3-carboxylic acid (4j)
Yellow solid, mp 186-187 ℃, yield 67%; 1H NMR (600 MHz, CDCl3): δH = 13.57 (s, 1H, COOH), 10.96 (s, 1H, OH), 8.83 (d, J = 2.4 Hz, 1H, H-6-py), 8.18 (d, J = 2.5 Hz, 1H, H-4-py), 7.89 (dd, J = 7.9, 3.0 Hz, 1H, H-Ar), 7.52-7.55 (m, 1H, H-Ar), 7.39 – 7.48 (m, 3H, H-Ar), 7.37 (d, J = 7.1 Hz, 1H, H-Ar), 7.29 – 7.24 (m, 1H, H-Ar), 7.07 – 7.00 (m, 1H, H-Ar), 5.32 (s, 2H, CH2N); 13C NMR (151 MHz, CDCl3): δC = 193.0, 163.9, 162.2, 161.1, 158.9, 154.7 (1JCF = 241.0 Hz), 147.0, 144.8, 133.3, 129.6, 128.8, 122.9 (2JCF = 25.4 Hz), 120.5 (3JCF = 7.5 Hz), 118.7, 117.7, 117.0, 116.2 (2JCF = 24.0 Hz), 54.0; HRMS-ESI (m/z) Calcd. for C20H15FNO5 [M+H]+: 368.0942; found, 368.0933.
1-allyl-5-(5-fluoro-2-hydroxybenzoyl)-2-oxo-1,2-dihydropyridine-3-carboxylic acid (4k)
Yellow solid, mp 122-123 ℃, yield 70%; 1H NMR (600 MHz, CDCl3): δH = 13.52 (s, 1H, COOH), 10.99 (s, 1H, OH), 8.83 (d, J = 2.1 Hz, 1H, H-6-py), 8.22 (d, J = 2.0 Hz, 1H, H-4-py), 7.30 (dt, J = 7.1, 2.8 Hz, 1H, H-Ar), 7.14 (dd, J = 8.3, 2.8 Hz, 1H, H-Ar), 7.07 (dd, J = 9.1, 4.4 Hz, 1H, H-Ar), 5.90-6.00 (ddt, J = 16.6, 10.2, 6.2 Hz, 1H, =CH), 5.48 (d, J = 10.2 Hz, 1H, =CH cis), 5.40 (d, J = 17.1 Hz, 1H, =CH trans), 4.78 (d, J = 6.1 Hz, 2H, CH2N); 13C NMR (151 MHz, CDCl3): δC = 193.1, 163.8, 163.7, 159.0, 154.8 (1JCF = 240 Hz), 145.5, 144.8, 129.9, 124.9 (2JCF = 23.6 Hz), 122.1 , 120.6 (3JCF = 6 Hz), 118.8, 117.7 (3JCF = 6 Hz), 117.5, 116.3 (2JCF = 24 Hz), 52.9; HRMS-ESI (m/z) Calcd. For Calcd. for C16H13FNO5 [M+H]+: 318.0547; found, 318.0505.
1-cyclohexyl-5-(5-fluoro-2-hydroxybenz oyl)-2-oxo-1,2-dihydropyridine-3-carboxylic acid (4l)
Yellow solid, mp 135-136 ℃, yield 79%; 1H NMR (600 MHz, CDCl3): δH = 13.77 (s, 1H, COOH), 11.01 (s, 1H, OH), 8.79 (s, 1H, H-6-py), 8.28 (s, 1H, H-4-py), 7.27 – 7.32 (dt, J = 7.1, 2.9 Hz, 1H, H-Ar), 7.14 (dd, J = 8.4, 2.9 Hz, 1H, H-Ar), 7.07 (dd, J = 9.1, 4.4 Hz, 1H, H-Ar), 4.91 – 4.98 (m, 1H, CH-cyclohexyl), 2.05 – 2.10 (m, 2H, H-cyclohexyl), 1.98 – 1.99 (m, 2H, H-cyclohexyl), 1.76– 1.82 (m, 2H, H-cyclohexyl), 1.52 – 1.58 (m, 4H, H-cyclohexyl); 13C NMR (151 MHz, CDCl3): δC = 193.5, 164.2, 163.7, 162.7, 158.9, 154.9 (1JCF = 241.0 Hz), 147.0, 144.0, 124.7 (2JCF = 23.5 Hz), 120.5 (3JCF = 6 Hz), 118.7, 117.8 (3JCF = 6 Hz), 116.3 (2JCF = 23.9 Hz), 57.3, 32.6, 25.6, 25.0;; HRMS-ESI (m/z) Calcd. for C19H19FNO5 [M+H]+: 360.1242; found, 360.1251.
5-(5-fluoro-2-hydroxybenzoyl)-2-oxo-1-(1-phenylethyl)-1,2-dihydropyridine-3-carboxylic acid (4m)
Yellow solid, mp 151-154 ℃, yield 63%; 1H NMR (300 MHz, CDCl3): δH = 13.64 (s, 1H, COOH), 10.99 (s, 1H, OH), 8.82 (d, J = 2.5 Hz, 1H, H-6-py), 8.03 (d, J = 2.5 Hz, 1H, H-4-py), 7.34 – 7.51 (m, 5H, H-Ar), 7.19 – 7.30 (m, 1H, H-Ar), 7.02 (dd, J = 9.1, 4.5 Hz, 1H, H-Ar), 6.83 (dd, J = 8.4, 2.9 Hz, 1H, H-Ar), 6.50 (q, J = 6.9 Hz, 1H, CHN), 1.87 (d, J = 7.0 Hz, 3H, CH3CH); 13C NMR (75 MHz, CDCl3): δC = 193.0, 163.9, 163.7, 158.9, 154.7(1JCF = 241 Hz), 144.4, 143.4, 137.4, 129.7, 129.6, 127.5, 124.6 (2JCF = 23.2 Hz), 120.4 (3JCF = 7.5 Hz), 118.6, 117.7, 117.6, 116.1(2JCF = 23.2 Hz), 56.1, 19.0; LCMS-ESI (m/z) Calcd. for C21H17FNO5 [M+H]+: 382.360 ; found, 382.2.
1-benzyl-5-(2-hydroxy-4-methoxybenzoyl)-2-oxo-1,2-dihydropyridine-3-carboxylic acid (4n)
Brown solid, mp 106-107 ℃, yield 57%; 1H NMR (300 MHz, CDCl3): δH = 13.42 (s, 1H, COOH), 12.04 (s, 1H, OH), 8.80 (d, J = 2.5 Hz, 1H, H-6-py), 8.19 (d, J = 2.5 Hz, 1H, H-4-py), 7.24 – 7.48 (m, 6H, H-Ar), 6.50 (d, J = 2.5 Hz, 1H, H-Ar), 6.42 (dd, J = 9.0, 2.5 Hz, 1H, H-Ar), 5.33 (s, 2H, CH2N), 3.86 (s, 3H, OMe); 13C NMR (75 MHz, CDCl3): δC = 192.1, 166.9, 166.2, 164.2, 163.9, 145.1, 145.0 , 133.7, 133.3, 129.5, 129.4, 128.7, 119.7, 117.2, 112.0, 108.3, 101.6, 55.8, 53.9; HRMS-ESI (m/z) Calcd. for C21H18NO6 [M+H]+: 380.1127; found, 380.1130.
5-(2-hydroxy-4-methoxybenzoyl)-2-oxo-1-vinyl-1,2-dihydropyridine-3-carboxylic acid (4o)
Brown solid, mp 103-106 ℃, yield 52%; 1H NMR (300 MHz, DMSO-d6): δH = 13.77 (s, 1H, COOH), 10.98 (s, 1H, OH), 8.65 (d, J = 2.6 Hz, 1H, H-6-py), 8.48 (d, J = 2.5 Hz, 1H, H-4-py), 7.46 (d, J = 8.6 Hz, 1H, H-Ar), 6.50 – 6.59 (m, 2H, H-Ar), 5.96 – 6.07 (m, 1H, CHCH2), 5.27 (d, J = 9.4 Hz, 1H, =CH cis), 5.23 (d, J = 16.4 Hz, 1H, =CH trans), 4.79 (d, J = 4.8 Hz, 2H, CH2N), 3.80 (s, 3H, -OMe); 13C NMR (75 MHz, DMSO-d6): δC = 190.5, 164.5, 164.4, 162.7, 160.3, 147.8, 144.4, 133.1, 131.9, 119.0, 118.9, 116.2, 115.8, 106.8, 101.3, 55.5, 52.0; HRMS-ESI (m/z) Calcd. for C17H16NO6 [M+H]+: 330.0670; found, 330.0664.
1-cyclohexyl-5-(2-hydroxy-4-methoxybenzoyl)-2-oxo-1,2-dihydropyridine-3-carboxylic acid (4p)
Brown solid, mp 158-161 ℃, yield 58%; 1H NMR (300 MHz, CDCl3): δH = 13.96 (s, 1H, COOH), 12.10 (s, 1H, OH), 8.78 (d, J = 2.2 Hz, 1H, H-6-py), 8.26 (d, J = 2.2 Hz, 1H, H-4-py), 7.42 (d, J = 8.8 Hz, 1H, H-Ar), 6.45 – 6.56 (m, 2H, H-Ar ), 4.84 – 5.14 (m, 1H, CH- cyclohexyl), 3.89 (s, 3H, OMe), 1.89 – 2.14 (m, 4H, H-cyclohexyl), 1.83 (m, 1H, H-cyclohexyl), 1.58 (m, 4H, H-cyclohexyl), 1.18 – 1.35 (m, 1H, H-cyclohexyl); 13C NMR (75 MHz, CDCl3): δC = 192.6, 166.9, 166.2, 164.5, 163.7, 144.2, 142.2, 142.1, 133.4, 119.7, 116.5, 112.1, 108.3, 101.7, 57.1, 55.8, 32.6, 25.6, 25.0; HRMS-ESI (m/z) Calcd. for C20H22NO6 [M+H]+: 372.1292; found, 372.1295.
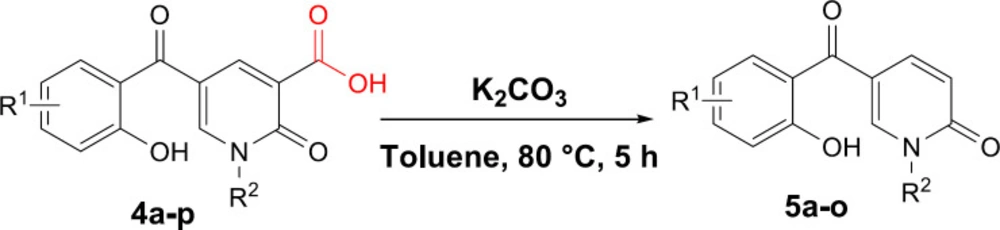
General procedure for the synthesis of 5a-o
5-(2-hydroxybenzoyl)-1-alkyl-2-oxo-1,2-dihydropyridine-3-carboxylic acid (0.2 mmol) and K2CO3 (0.4 mmol,55 mg) were added to an oven-dried flask containing toluene (0.5 mL) and the mixture was refluxed for 8 h. After completing the reaction based on TLC, the mixture was cooled to room temperature. The solvent was evaporated in vacuo, then DCM (15 mL) was added to the crude product and extracted from water (3 × 15 mL). The organic layer was dried over Na2SO4 and evaporated under reduced pressure. The crude product was purified by column chromatography using hexane/EtOAc 3:1 as eluent.
1-benzyl-5-(2-hydroxybenzoyl)pyridin-2(1H)-one (5a)
White crystal, mp 120-121 ℃, yield 86%; 1H NMR (400 MHz, CDCl3): δH = 11.43 (s, 1H,OH), 7.92 (d, 1H, J = 2.2 Hz, H-6-py), 7.76 (dd, 1H, J = 9.4, 2.2 Hz, H-4-py), 7.48 (t, 1H, J = 7.7 Hz, H-Ar), 7.32 – 7.42 (m, 6H, H-Ar), 7.04 (d, 1H ,J = 8.3 Hz, H-Ar), 6.83 (t, 1H, J = 7.5 Hz, H-Ar), 6.66 (d, 1H, J = 9.5 Hz, H-3-py), 5.19 (s, 2H, –CH2N); 13C NMR (100 MHz, CDCl3): δC = 195.1, 162.4, 162.1, 143.2, 139.0, 136.2, 135.3, 131.5, 129.2, 128.6, 128.5, 120.2, 118.8, 118.9, 118.7, 117.3, 52.6; HRMS-ESI (m/z) Calcd. for C19H16NO3 [M+H]+: 306.1135; found, 306.1138.
1-allyl-5-(2-hydroxybenzoyl)pyridin-2(1H)-one (5b)
White crystal, mp 128-129 ℃, yield 88%; 1H NMR (300 MHz, CDCl3): δH = 11.43 (s, 1H, OH), 7.93 (d, 1H, J = 2.3 Hz, H-6-py), 7.75 (dd, 1H, J = 9.5, 2.6 Hz, H-4-py), 7.44 – 7.59 (m, 2H, , H-Ar), 7.08 (d, 1H, J = 8.3 Hz, H-Ar), 6.93 (t, 1H, J = 7.6 Hz, H-Ar), 6.64 (d, 1H, J = 9.5 Hz, H-3-py), 5.91 – 6.06 (m, 1H, CHCH2), 5.36 (d, 1H, J = 10.1 Hz, CH2=CH cis), 5.29 (d, 1H, J = 17.1 Hz, CH2=CH trans), 4.64 (d, 2H, J = 5.8 Hz, CH2N); 13C NMR (75 MHz, CDCl3): δC = 195.1, 162.5, 161.7, 142.9, 139.0, 136.5, 131.5, 120.1, 120.0, 119.8, 118.9, 118.8, 118.7, 117.3, 51.7; HRMS-ESI (m/z) Calcd. for C15H14NO3 [M+H]+: 256.0960; found, 256.0964.
1-cyclohexyl-5-(2-hydroxybenzoyl)pyridin-2(1H)-one (5c)
Yellow crystal, mp 115-116 ℃, yield 83%; 1H NMR (300 MHz, CDCl3): δH = 11.49 (s, 1H, OH), 8.00 (d, 1H, J = 2.5 Hz, H-6-py), 7.73 (dd, 1H, J = 9.5, 2.6 Hz, H-4-py), 7.50 – 7.60 (m, 2H, H-Ar), 7.08 (d, 1H, J = 7.8 Hz, H-Ar), 6.95 (dt, 1H, J = 8.4, 1.2 Hz, H-Ar), 6.61 (d, 1H , J = 9.5 Hz, H-3-py), 4.91 (m, 1H, CH, CH- cyclohexyl), 2.04 – 1.36 (m, 10H, H-cyclohexyl);13C NMR (75 MHz, CDCl3): δC = 195.4, 162.5, 161.7, 139.9, 138.1, 136.0, 131.5, 119.6, 118.9, 118.8, 118.7, 117.1, 54.8, 32.6, 25.7, 25.2; HRMS-ESI (m/z) Calcd. for C18H20NO3 [M+H]+: 298.1540; found, 298.1533.
1-(furan-2-ylmethyl)-5-(2-hydroxybenzoyl)pyridin-2(1H)-one (5d)
Yellow solid, mp 127-128 ℃, yield 68%; 1H NMR (400 MHz, CDCl3): δH = 11.46 (s, 1H, OH), 7.99 (d, 1H ,J = 2.2 Hz, H-6-py), 7.77 (dd, 1H, J = 9.5, 2.4 Hz, H-4-py), 7.50 (t, 2H, J = 8.0 Hz, H-Ar), 7.43 (d, 1H, J = 1.8 Hz, H-furyl ), 7.07 (d, 1H, J = 8.2 Hz, H-Ar), 6.90 (t, 1H, J = 7.6 Hz, H-Ar), 6.64 (d, 1H, J = 9.5 Hz, H-3-py), 6.51 (d, 1H, J = 3.0 Hz, H-furyl), 6.36 – 6.42 (m, 1H, H-furyl), 5.18 (s, 2H, CH2N);13C NMR (100 MHz, CDCl3): δC =195.1, 162.5, 161.7, 147.8, 143.6, 142.9, 139.1, 136.2, 131.7, 120.2, 118.9, 118.8, 118.7, 117.3, 111.1, 111.0, 45.1; HRMS-ESI (m/z) Calcd. for C17H14NO4 [M+H]+: 296.0931; found, 296.0921.
5-(2-hydroxybenzoyl)-1-(prop-2-yn-1-yl)pyridin-2(1H)-one(5e)
White solid, mp 117-116 ℃, yield 68%; 1H NMR (400 MHz, CDCl3): δH = 11.46 (s, 1H, OH), 8.35 (d, 1H, J = 2.4 Hz , H-6-py), 7.82 (dd, 1H , J = 9.5, 2.5 Hz, H-4-py), 7.56 – 7.62 (m, 1H, H-Ar), 7.56 – 7.49 (m, 1H, H-Ar), 7.09 (d, 1H, J = 8.3 Hz,H-Ar), 6.94 (t, 1H, J = 7.5 Hz, H-Ar), 6.65 (d, 1H , J = 9.5 Hz, H-3-py), 4.83 (d, 2H, J = 2.5 Hz, CH2N), 2.61 (t, 1H, J = 2.5 Hz, CH≡C); 13C NMR (100 MHz, CDCl3): δC = 195.1, 162.4, 162.1, 141.7, 139.4, 136.7, 136.3, 131.6, 129.0, 119.7, 118.9, 118.8, 117.5, 75.8, 38.5; HRMS-ESI (m/z) Calcd. for C15H12NO3 [M+H]+: 254.0610; found, 254.0601.
5-(2-hydroxybenzoyl)-1-(1-phenylethyl)pyridin-2(1H)-one (5f)
Yellow solid, mp 95-97 ℃, yield 87%; 1H NMR (400 MHz, CDCl3): δH = 11.42 (s, 1H, OH), 7.77 (d, 1H, J = 2.5 Hz, , H-6-py), 7.70 – 7.73 (m, 1H , H-4-py ), 7.33 – 7.48 (m, 6H, H-Ar), 7.11 (dd, 1H , J = 8.0, 1.4 Hz, H-Ar), 7.01 (d, 1H , J = 8.0 Hz, H-Ar), 6.68 (t, 2H, J = 8.0 Hz, H-Ar, H-3-py ), 6.46 (q, 1H, J = 7.0 Hz, CHN), 1.74 (d, 3H, J = 7.0 Hz, CH3); 13C NMR (100 MHz, CDCl3): δC = 194.9, 162.4, 161.8, 141.1, 139.2, 138.2, 135.9, 131.3, 129.2, 128.5, 127.5, 120.0, 118.7, 118.6, 118.5, 117.1, 53.7, 18.9; HRMS-ESI (m/z) Calcd. for C20H18NO3 [M+H]+: 320.1393; found, 320.1397.
1-benzyl-5-(5-fluoro-2-hydroxybenzoyl)pyridin-2(1H)-one (5j)
Yellow solid, mp 105-106 ℃, yield 84%; 1H NMR (300 MHz, CDCl3): δH = 11.08 (s, 1H, OH), 7.93 (d, 1H, J = 2.4 Hz, H-6-py), 7.76 (dd, 1H, J = 9.5, 2.6 Hz, , H-4-py), 7.32 – 7.45 (m, 5H, H-Ar), 7.19 – 7.27 (m, 1H, H-Ar), 7.08 (dd, 1H, J = 8.7, 3.0 Hz, H-Ar), 7.02 (dd, 1H, J = 9.1, 4.5 Hz, H-Ar), 6.68 (d, 1H, J = 9.5 Hz, , H-3-py), 5.20 (s, 2H, CH2N); 13C NMR (75 MHz, CDCl3): δC = 194.0, 161.9, 158.5, 154.6 (1JCF = 240.0 Hz), 143.2, 138.5, 135.1, 129.3, 128.7, 128.5, 123.5 (2JCF = 23.6 Hz), 120.4, 120.0 (3JCF = 6.0 Hz), 118.3 (3JCF = 6.1 Hz), 116.8, 116.4 (2JCF = 24.0 Hz), 52.6; HRMS-ESI (m/z) Calcd. for C19H15FNO3 [M+H]+: 324.1014; found, 324.1012.
1-allyl-5-(5-fluoro-2-hydroxybenzoyl)pyridin-2(1H)-one (5k)
Yellow solid, mp 100-101 ℃, yield 78%; 1H NMR (300 MHz, CDCl3): δH = 11.12 (s, 1H, OH), 7.96 (d, 1H , J = 2.5 Hz, H-6-py), 7.77 (dd, 1H, J = 9.5, 2.5 Hz, H-4-py), 7.16 – 7.33 (m, 2H, H-Ar), 7.06 (dd, 1H, J = 8.9, 4.6 Hz, H-Ar), 6.66 (d, 1H, J = 9.5 Hz, H-3-py), 5.89 – 6.09 (m, 1H, CHCH2), 5.35 – 5.41 (m, 1H, =CH cis), 5.30 (dd, 1H, J = 16.0, 0.9 Hz, =CH trans), 4.65 (d, 2H, J = 6.0 Hz, CH2N); 13C NMR (75 MHz, CDCl3): δC = 194.1, 161.6, 158.5, 154.7 (1JCF = 239.5 Hz), 143.1, 138.6, 131.5, 131.4, 123.6 (2JCF = 23.7 Hz), 120.2, 120.1 (3JCF = 6.5 Hz), 118.4 (3JCF = 6.4 Hz), 116.8, 116.5 (2JCF = 24.3 Hz), 51.7; HRMS-ESI (m/z) Calcd. for C15H13FNO3 [M+H]+: 274.0825; found, 274.0820.
1-cyclohexyl-5-(5-fluoro-2-hydroxybenz-oyl)pyridin-2(1H)-one (5l)
Yellow solid, mp 125-126 ℃, yield 78%; 1H NMR (300 MHz, CDCl3): δH = 11.17 (s, 1H, OH), 8.02 (d, 1H, J = 2.5 Hz, H-6-py), 7.72 (dd, 1H, J = 9.5, 2.6 Hz, H-4-py), 7.18 – 7.34 (m, 2H, H-Ar), 7.06 (dd, 1H, J = 9.0, 4.6 Hz, H-Ar), 6.62 (d, 1H, J = 9.5 Hz, H-3-py), 4.81 – 4.98 (m, 1H, CH- cyclohexyl), 1.11 – 2.07 (m, 10H, H-cyclohexyl); 13C NMR (75 MHz, CDCl3): δC = 194.3, 161.6, 158.6, 158.5, 154.7 (1JCF = 239.6 Hz), 140.1, 137.8, 123.4 (2JCF = 23.6 Hz), 120.1 (3JCF = 6.0 Hz), 119.7, 118.5 (3JCF = 6.3 Hz), 116.5 (2JCF = 23.8 Hz), 54.9, 32.6, 25.7, 25.2; HRMS-ESI (m/z) Calcd. for C18H19FNO3 [M+H]+: 316.1212; found, 316.1207.
1-benzyl-5-(2-hydroxy-4-methoxybenzoyl)pyridin-2(1H)-one (5o)
Brown solid, mp 83-85 ℃, yield 60%; 1H NMR (400 MHz, CDCl3): δH = 12.20 (s, 1H, OH), 7.86 (d, 1H, J = 2 Hz, H-6-py), 7.71 (dd, 1H, J = 9.5, 2.2 Hz, H-4-py), 7.29 – 7.43 (m, 6H, H-Ar), 6.66 (d, 1H, J = 9.5 Hz, H-3-py), 6.49 (d, 1H, J = 2.1 Hz, H-Ar), 6.37 (dd, 1H , J = 8.9, 2.2 Hz, H-Ar), 5.20 (s, 2H, CH2N), 3.85 (s, 3H, OCH3); 13C NMR (100 MHz, CDCl3): δC= 193.7, 166.2, 165.8, 162.1, 142.2, 139.0, 135.4, 133.3, 129.2, 128.6, 128.5, 120.2, 117.7, 112.5, 107.6, 101.4, 55.7, 52.5; HRMS-ESI (m/z) Calcd. for C20H18NO4 [M+H]+: 336.1015; found, 336.1010.
General procedure for the synthesis of 1-benzyl-2-oxo-1,2-dihydropyridine-3-carbo-xylic acid (6) (36)
Compound 2-chloro-1,2-dihydropyridine-3-carboxylic acid (1 mmol) in 70% aqueous acetic acid (10 mL) was heated under reflux for 4-6 h (see ref 5 in article). Completion of the reaction was checked by TLC. After cooling a solid product precipitated which was filtered, washed well with water, dried and purified by recrystallization from DMF. Compound 2-oxo-1,2-dihydropyridine-3-carboxylic acid (1 equiv) in DMF (4 mL) BnBr (1.3 equiv) and potassium carbonate (1.5 equiv) were added. The reaction mixture was stirred at room temperature during 20-50 h. After completion (checked by TLC) the reaction mixture was poured into ice-water (25 mL) and the solid product formed was filtered, washed well with water, dried and purified by recrystallization from EtOH.
1-benzyl-2-oxo-1,2-dihydropyridine-3-carboxylic acid (6)
White solid, mp 128-130 ℃, yield 67%; 1H NMR (300 MHz, DMSO-d6): δH = 14.46 (s, 1H, COOH), 8.42-8.36 (m, 2H, H-4,6-py), 7.40-7.27 (m, 5H, Bn), 6.76 (t, J = 6.8 Hz, 1H, H-5-py), 5.31 (s, 2H, -CH2); 13C NMR (75 MHz, , DMSO-d6): δC = 164.72, 163.6, 145.6, 145.1, 135.8, 128.8, 128.1, 127.9, 116.9, 108.8, 52.6.
General procedure for the preparation of Methyl 1-benzyl-5-(2-hydroxybenzoyl)-2-oxo-1,2-dihydropyridine-3-carboxylate(7)
Compound 4a (1mmol) in DMF (4 mL) were added methyl iodide (1 mmol) and potassium carbonate (1.5 mmol). The reaction mixture was stirred at rt for 2.5 days. Water was added and the reaction mixture was extracted with hexanes-ether (1:1) solvent system. The combined organic phases were dried over Na2SO4, filtered, and concentrated in a vacuum to yield 8a (40%) as an orange solid.
Red solid, mp 150-152 ℃, yield 60%; 1HNMR (300 MHz, CDCl3): δH = 8.49 (d, J = 2.6 Hz, 1H, H-6-py), 8.16 (d, J = 2.6 Hz, 1H, H-4-py), 7.45 (t, J = 7.5 Hz, 1H, H-Ar), 7.36-7.29 (m, 6H, H-Ar), 7.01 (t, J = 7.5 Hz, 1H, H-Ar), 6.91 (d, J = 8.4 Hz, 1H, H-Ar), 5.15 (s, 2H, CH2N), 3.85 (s, 3H, CH3), 3.60 (s, 3H, CH3), 13CNMR (75 MHz, CDCl3): δC = 190.4, 165.0, 159.0, 156.6, 147.3, 144.2, 134.9, 132.8, 129.8, 129.0, 128.5, 126.8, 121.1, 119.3, 116.8, 111.3, 55.4, 53.3, 53.4; HRMS-ESI (m/z) C22H20NO5 [M+H]+ : 378.1341; found, 378.1349.
Ethyl 1-benzyl-5-(2-hydroxybenzoyl)-2-oxo-1,2-dihydropyridine-3-carboxylate (8)
White solid, mp 115-117 ℃, yield 73%; 1HNMR (600 MHz, CDCl3): δH = 11.38 (s, 1H, OH), 8.53 (s, 1H, H-6-py), 8.14 (s, 1H, H-4-py), 7.53 (t, J = 7.6 Hz, 1H, H-Ar), 7.43 – 7.37 (m, 6H, H-Ar), 7.09 (d, J = 8.4 Hz, 1H, H-Ar), 6.88 (t, J = 7.7 Hz, 1H, H-Ar), 5.26 (s, 2H, CH2N), 4.42 (q, J = 7.2 Hz, 2H, CH2O), 1.41 (t, J = 7.2 Hz, 3H, CH3CH2); 13CNMR (151 MHz, CDCl3): δC = 194.3, 164.1, 162.6, 158.7, 146.1, 143.6, 136.5, 134.8, 131.4, 129.3, 128.9, 128.8, 120.5, 119.0, 118.9, 118.5, 115.9, 61.7, 53.3; HRMS-ESI (m/z) C22H20NO5 [M+H]+ : 378.1278; found, 378.1273.
Antimicrobial assay
All new structures were assessed to in vitro tests for their antibacterial properties. The antibacterial activities were evaluated against E. coli PTCC 25922, A. baumannii (clinical strain); S. aureus ATCC 1431; C. albicans ATCC 10231 and Methicillin-resistant S. aureus (clinical strain).
The values of minimum inhibitory concentration (MIC) were assayed by the standard technique described by CLSI (
37) meaning standard broth micro-dilution procedure.
The culture of each strain was prepared on a nutrient agar plate and remained overnight, then, it was used to make the bacterial suspensions equal to 0.5 McFarland standards in sterile normal saline. Then Serial dilutions of samples were obtained in sterile 96 wells plates containing Mueller–Hinton broth which was prepared with concentrations ranging from 0.031 to 64 mg/ml. The final concentration of bacterial cells in each well was 0.5–1 ×106 cfu mL-1 approximately.
Minimum inhibitory concentrations (MICs) were recorded after 22 hours of incubation at 37 ºC. Each experiment was performed in triplicate. Cefixime, Ciprofloxacin and Nystatin were used as the reference antibiotics for bacteria and yeast, respectively.
Molecular modeling procedure
Target enzyme and ligand preparation
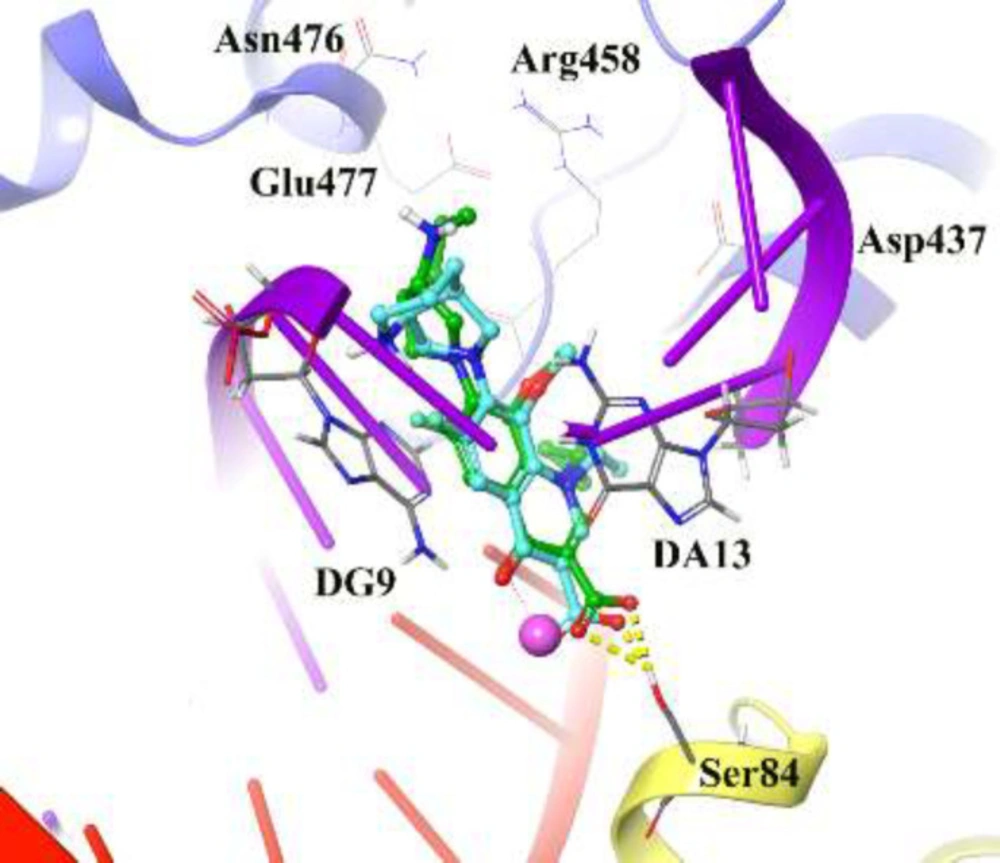
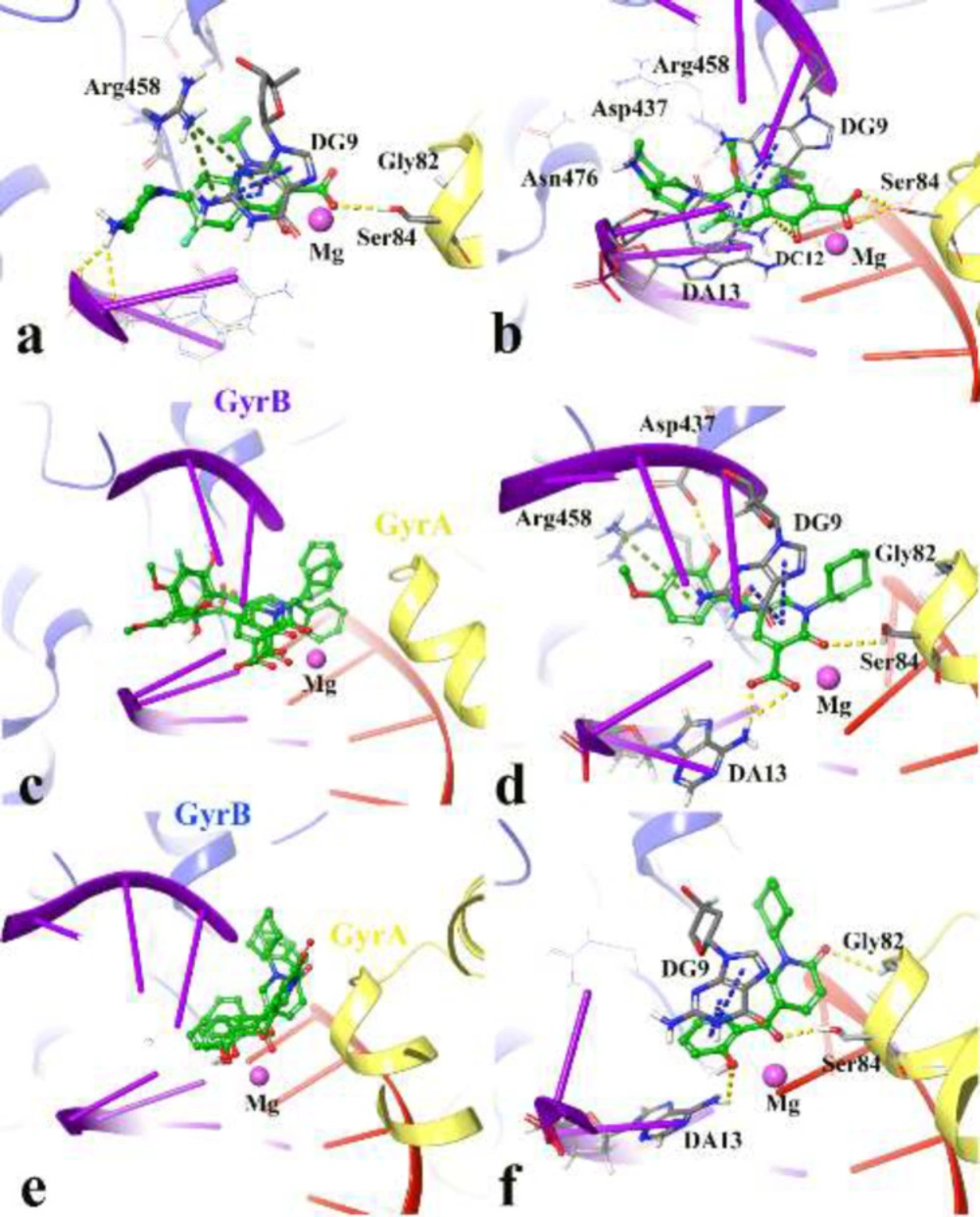
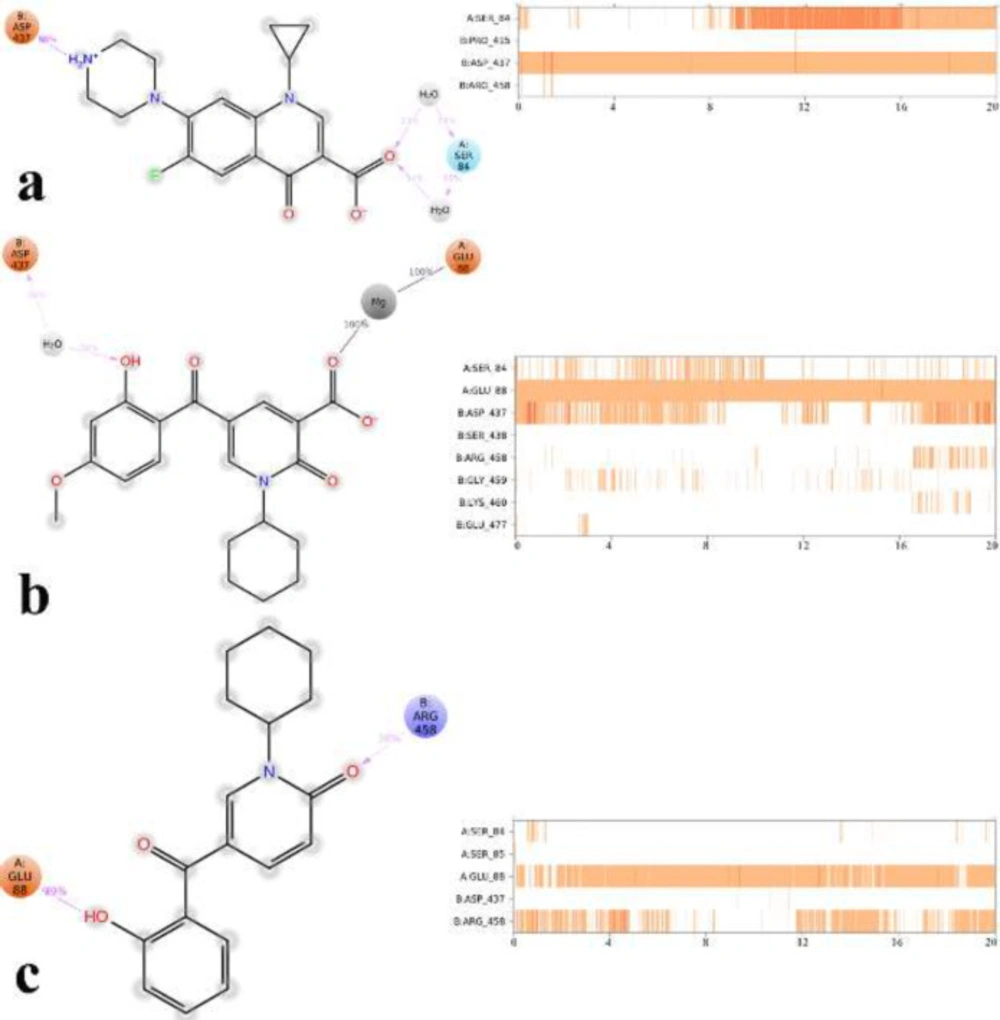
To find out the interactions mode of designed molecules over DNA gyrase, the Maestro Molecular Modeling platform (version 10.5) by Schrödinger, LLC was performed (38). The X-ray crystallographic structure of
S. aureus DNA gyrase (in complex with moxifloxacin and DNA) was downloaded from the Protein Data Bank (PDB ID; 5cdq) (www.rcsb.org) (
39).
As urease is reported to be functionally active in a monomeric state, all the docking studies were performed on a single monomer. In addition, prosthetic group and co-factors are not directly involved in urease inhibition, so they were removed before docking investigation. Water molecules and co-crystallized ligands were removed from the enzyme’s crystallographic structures.
The 2D structures of all synthesized compounds were drawn in Marvin 15.10.12.0 program (http://www.chemaxon.com) (
40) and converted into a pdb file. The Protein Preparation Wizard (
41) and the LigPrep (
42) module were used to prepare protein and ligand structure properly. The missing side chains of the proteins were filled using the Prime tool and missing residues were updated.
Induced fit docking (IFD) protocol
IFD method implemented in Glide software (Schrödinger LLC 2018, USA) was used to investigate the possible binding mode of the most active inhibitors in the active site of DNA gyrase (
38). The moxifloxacin binding site was used to generate the grid for IFD calculation. The maximum 20 poses with receptor and ligand van der Waals radii of 0.7 and 0.5, respectively considered. Residues within 5 Å of the moxifloxacin at the active site were refined followed by side-chain optimization. Structures whose Prime energy is more than 30 kcal/mol are eliminated based on extra precious Glide docking.
Molecular dynamic (MD) simulation
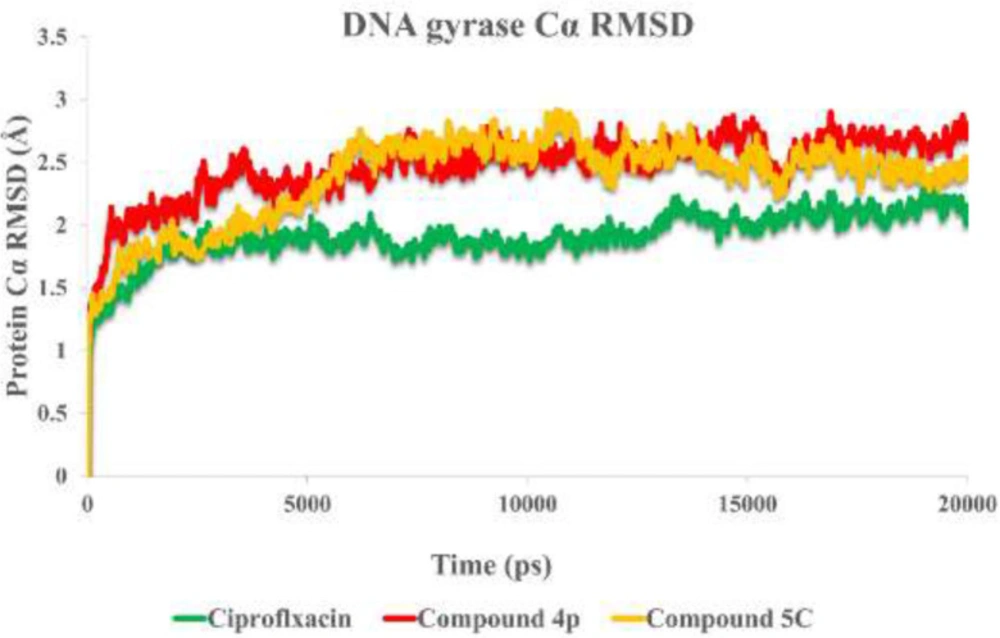
Molecular simulations of this study were performed using the Desmond v5.3 using the Maestro interface (from Schrödinger 2018‐4 suite) (
43). The appropriate pose for the MD simulation procedure of the compound was achieved by the IFD method. To build the system for MD simulation, the protein-ligand complexes were solvated with SPC explicit water molecules and placed in the center of an orthorhombic box of appropriate size in the periodic boundary condition. Sufficient counter‐ions and a 0.15 M solution of NaCl were also utilized to neutralize the system and to simulate the real cellular ionic concentrations, respectively. The MD protocol involved minimization, pre-production, and finally production MD simulation steps. In the minimization procedure, the entire system was allowed to relax for 2500 steps by the steepest descent approach. Then the temperature of the system was raised from 0 to 300 K with a small force constant on the enzyme to restrict any drastic changes. MD simulations were performed via NPT (constant number of atoms, constant pressure
i.e. 1.01325 bar and constant temperature
i.e. 300 K) ensemble. The Nose‐Hoover chain method was used as the default thermostat with 1.0 ps interval and Martyna‐Tobias‐Klein as the default barostat with 2.0 ps interval by applying an isotropic coupling style. Long‐range electrostatic forces were calculated based on the particle‐mesh‐based Ewald approach with the cut‐off radius for columbic forces set to 9.0 Å. Finally, the system was subjected to produce MD simulations for 20 ns for each protein-ligand complex. During the simulation was stored every 1000 ps of the actual frame. The dynamic behavior and structural changes of the systems were analyzed by the calculation of the root mean square deviation (RMSD) and RMSF. Subsequently, the energy-minimized structure calculated from the equilibrated trajectory system was evaluated for the investigation of each ligand-protein complex interaction.
In-silico ADME properties of synthesized compounds
QikProp module of Schrodinger was applied to calculate the important pharmacokinetic properties of the synthesized compounds like drug-likeness, metabolism and cell permeation (
44).