Materials
DMEM-F12 (Gibco, USA), DMEM (Sigma-Aldrich, USA), Trypsin-EDTA (Sigma-Aldrich, USA), Fetal bovine serum (Gibco, USA), EDTA (Sigma-Aldrich, USA), Penicillin–Streptomycin Solution (Sigma-Aldrich, USA), Antibiotic–antimycotic (Invitrogen, USA), PBS (Gibco, USA), Phenol red (Sigma-Aldrich, USA), Trypan blue (Sigma-Aldrich, USA), MTT powder (SRL, India), DMSO [Dimethyl sulfoxide] (Sigma-Aldrich, USA), Triton X-100 (Sigma-Aldrich, USA), Agarose (Sigma-Aldrich, USA), Ethidium bromide (Merc, Germany), Griess reagent (Sigma-Aldrich, USA), Flat Bottom 96-well plate (Sigma-Aldrich, USA), Flat Bottom 24-well plate (Sigma-Aldrich, USA).
Scorpion venom preparation
Scorpions were collected from Kazeron in Fars province. Venom was collected by electric shock in telson region of scorpion tails. Venom then was frozen in -50 °C and lyophilized. The lyophilized powder was stored in -20 °C for future use. The powder then was solubilized in DMEM medium (without phenol red) and protein concentration was measured using Bradford method (
14).
Cell culture
MCF-7 and Vero cell lines were purchased from Iranian biological resource center. The cells were cultured in 75 mL flasks contained DMEM and 10% (v/v) FBS and 10 μL/mL penicillin-streptomycin antibiotics. The cells were incubated in 37 °C, 5% (v/v) CO2 and 80% (v/v) humidity (optimum conditions) and media were discarded three times a week. After appropriate confluence, the cells were trypsinized by Trypsin-EDTA and were counted by hemocytometer.
Cell treatment
The cells were seeded in 96 and 24-well plate and were incubated overnight in the CO2 incubator. After a night of incubation, the media was discarded and the fresh media containing the different concentration (25-200 μg/mL) of crude venom was added to every well. For each test cells were seeded in 96 or 24-well plate and incubated overnight in 5% (v/v) CO2, 80% (v/v) humidity, and 37 °C. Then the old media was discarded and new media containing 25, 50, 100, and 200 μg/mL of venom were added and incubated for 24 h in 37 °C. Control group was not treated with venom.
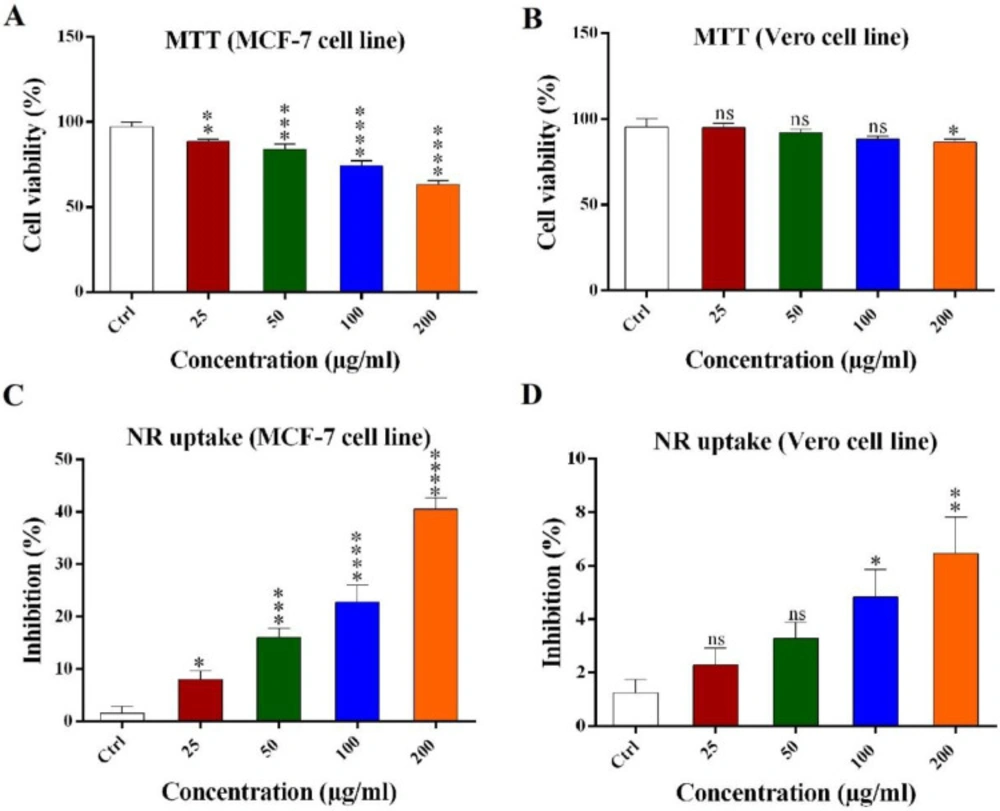
MTT reduction assay
The viability effect of scorpion venom was measured by the colorimetric MTT reduction assay (
15). 2 × 10
4 cells were counted by hemocytometer and seeded in 96-well plate and incubated under aforementioned conditions. Then MTT dye with final concentration of 5 mg/mL was added and incubated for 1hr in dark condition at 37 °C. Then, 200 μL DMSO was added to every well and incubated 2 h in dark condition. The absorbance was measured at 595 nm.
Neutral red (NR) uptake assay
NR uptake assay was used to estimate the number of viable cells and to confirm the MTT assay results (
16). 2 × 10
4 cells were seeded in 96-well plate and incubated under aforementioned conditions. Amount of 20 μL neutral red dye (5 μg/mL) was added to every well and incubated for 1 h in 37 °C. After red crystals formation, the supernatant was discarded and washed with PBS two times. One-hundred microliter fixation buffer (formaldehyde 37% (v/v), CaCl
2 10% (v/v), water) was added to every well and incubated for 1 min and then one-hundred microliter solubilizing buffer (acetic acid 5%) was added and incubated for 20 min in dark condition in a shaker incubator. The absorbance was measured at 540 nm.
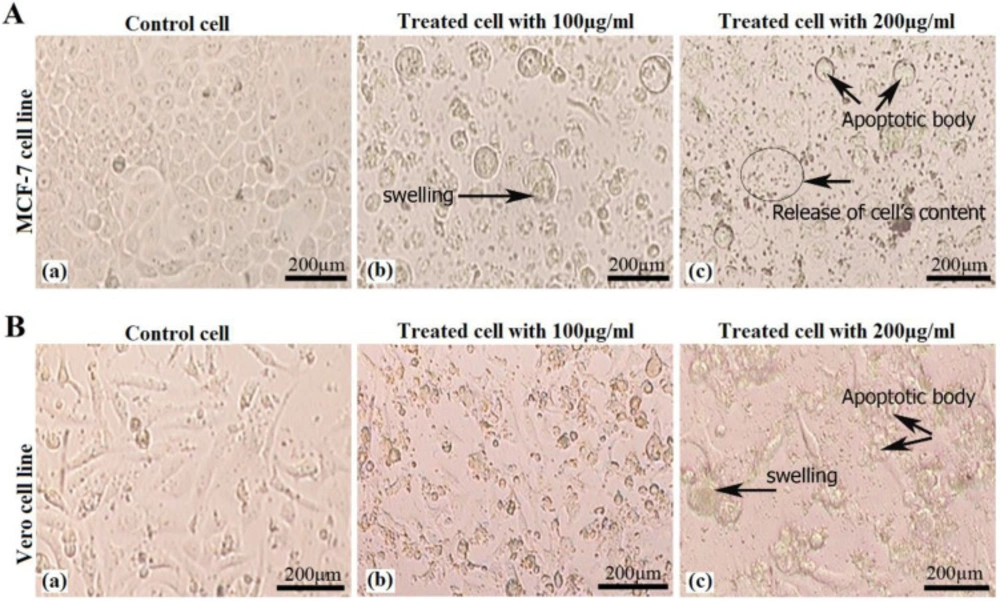
Cells morphology evaluation
2 × 104 cells were seeded in 96-well plate containing one-hundred microliter media (without serum) and incubated overnight in optimum conditions. Then old media was discarded and new media containing 100 and 200 μg/mL of venom were added and incubated for 24 h at 37 °C. Cells morphology was analyzed and some pictures were captured using inverted microscope (Nikon, Japan).
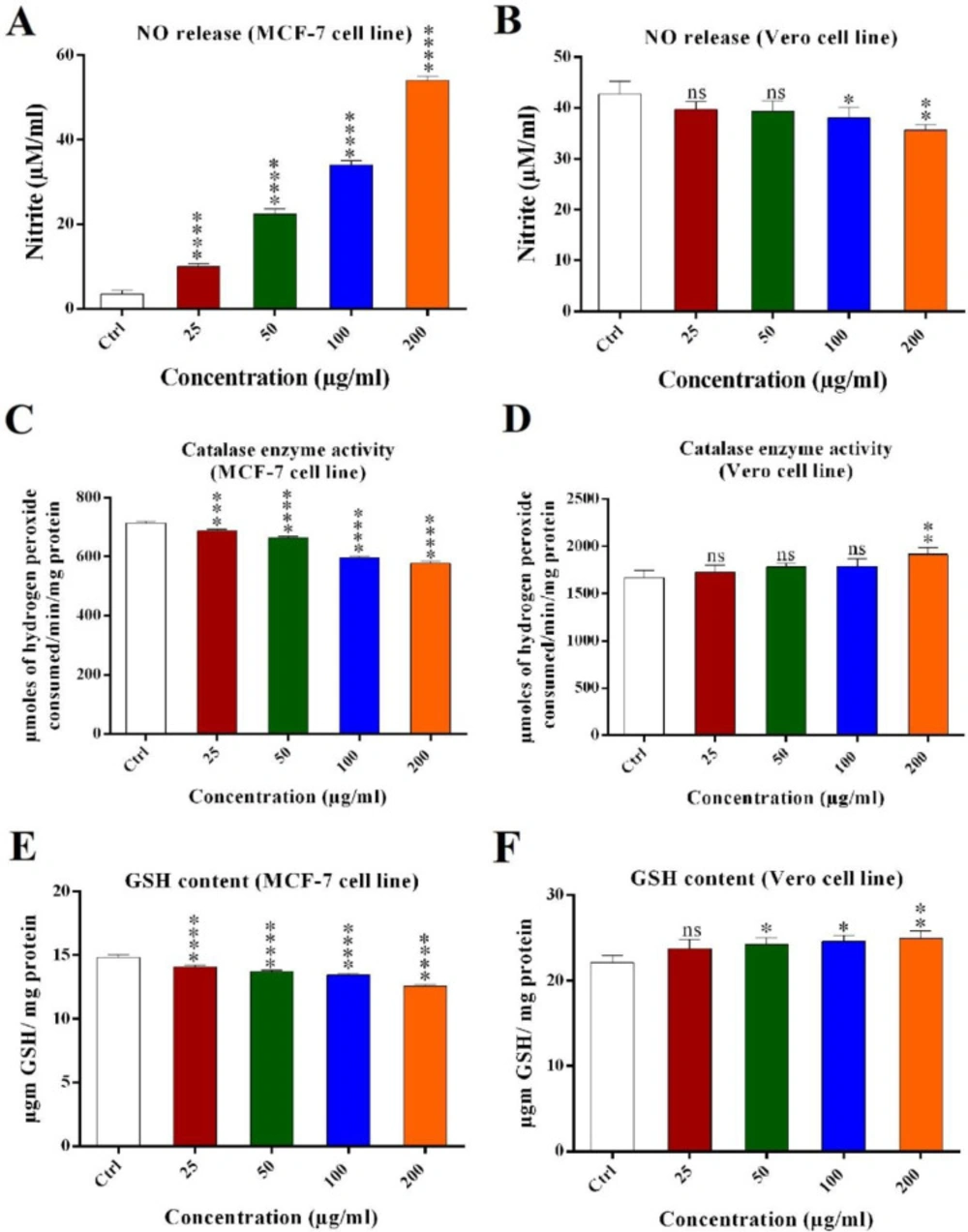
Nitric oxide (NO) assay
Nitric oxide was determined by measuring the nitrite content in culture medium (
17). 2 × 10
4 cells were seeded in 96-well plate and incubated under aforementioned conditions. Then media was transferred to fresh tube and centrifuged in 500 rpm for 5 min in 4 °C. One-hundred microliter media was transferred to fresh 96-well plate and mixed with equal volume of Griess reagent (0.04 g/mL PBS, pH 7.4) and incubated for 10 min at room temperature. Absorbance was measured in 540 nm by a micro plate reader (Biorad, USA). Nitrite oxide concentration (μM/mL) in treated and untreated cells was calculated using nitrite oxide standard curve.
Reduced glutathione (GSH) assay
Total reduced glutathione (GSH) was measured following the method of Sedlak and Linsay (
18). 5 × 10
5 cells were seeded in 24-well plate and incubated under aforementioned conditions. The cells then were trypsinized and harvested in fresh 1.5 mL tubes and centrifuged in 1500 rpm for 5 min in 4 °C and washed with PBS (pH 7.4) two times and incubated in -20 °C for 30 min. Two-hundred microliter chilled lysis buffer (NaCl 2.5M, EDTA 100 mM, Tris 10 mM, NaOH 0.2M, Triton X-100 1% and pH 10) was added and incubated for 30 min at room temperature. After 10-15 min sonication, the cells suspension was centrifuged in 2000 rpm for 10 min and a supernatant was transferred to the fresh tubes. The protein concentration was measured using Bradford method and an equal volume of 10% (v/v) TCA was added and stored at 4 °C for 2 h and then centrifuged and a supernatant was transferred to the fresh tubes. Twenty microliter of a supernatant was mixed with 75 μL of lysis buffer, 55 μL of Tris buffer (pH 8.5) containing 0.02M EDTA and 25 μL DTNB (5, 5/dithiobis (2-N benzoic acid)). The absorbance of yellow chromogen was measured using microplate reader. The result was expressed as μg GSH/mg protein using molar extinction coefficient of 13600.
Catalase enzyme activity assay
Catalase enzyme activity was measured using cells extract (
19). The number of 5 × 10
5 cells were seeded in 24-well plate and incubated under aforementioned conditions. Then, the cells were trypsinized and harvested in fresh 1.5 mL tubes and centrifuged in 1500 rpm for 5 min in 4 °C and washed with PBS (pH 7.4) two times and incubated in -20 °C for 30 min. Two-hundred microliter chilled lysis buffer (NaCl 2.5M, EDTA 100 mM, Tris 10 mM, NaOH 0.2 M, Triton X-100 1% and pH 10) was added and incubated for 30 min at room temperature. After 10-15 min sonication, the cell suspension was centrifuged in 2000 rpm for 10 min and a supernatant was transferred to the fresh tubes. The protein concentration was measured using Bradford method and 5 μL of the samples was mixed with 50 μL lysis buffer, 20 μL DDW, and 25 μL H
2O
2 (15%). The samples were incubated at 37 °C for 2 min and were mixed with one-hundred microliter dichromate acid reagent (0.1 M potassium dichromate in glacial acetic acid) then incubated in boiling water 10-15 min until greenish or faint greenish color was seen. Two-hundred microliter of the samples were transferred to flat 96-well plate and the absorbance was measured in 570 nm using the plate reader. The results were converted into the activity using molar extinction of Catalases (43.6) and expressed as micromoles of hydrogen peroxide consumed/min/mg protein.
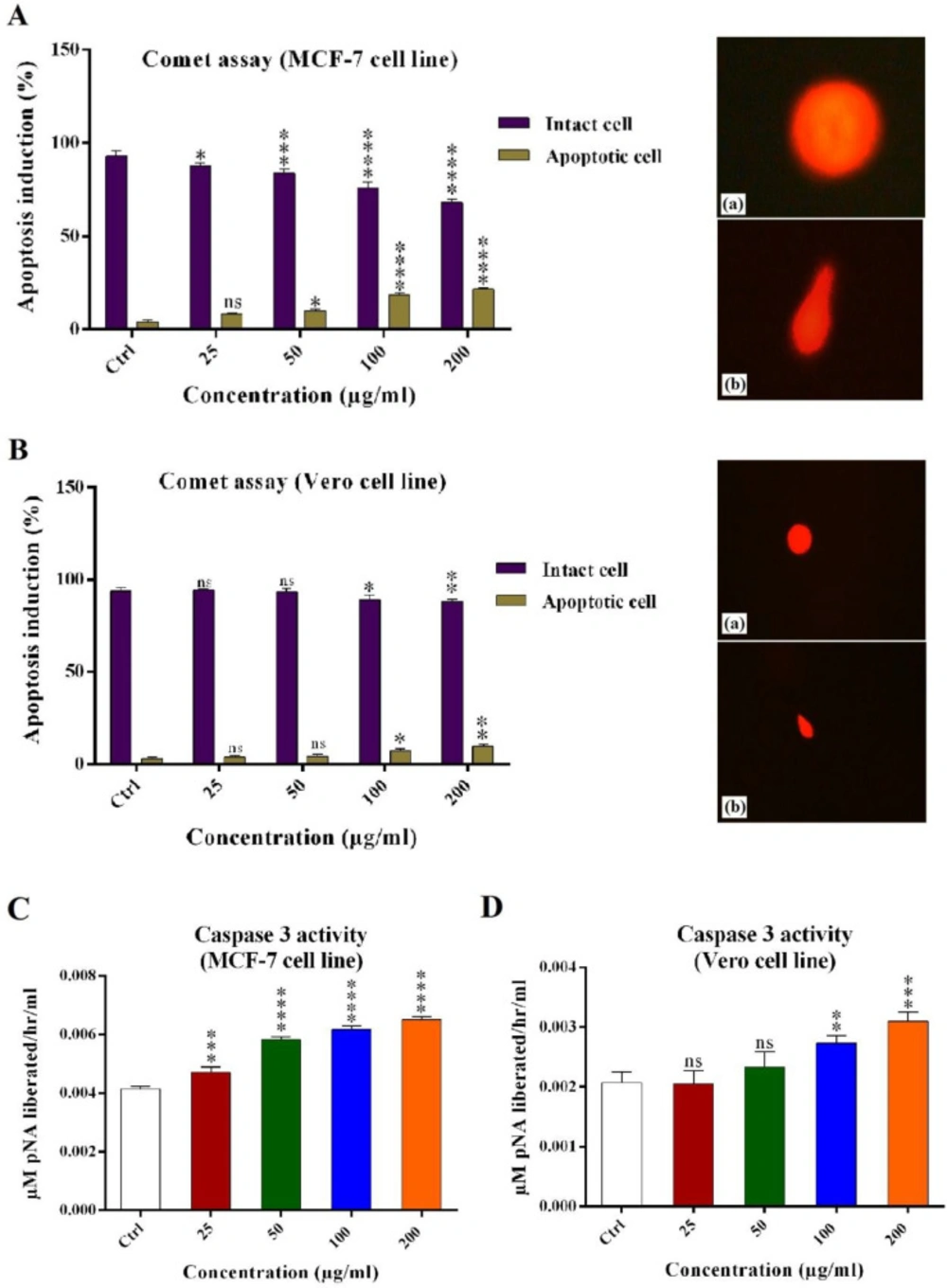
Alkaline comet assay
Alkaline comet or single-cell gel electrophoresis assay is a good method for DNA fragmentation analysis in the cells (
20). 12 × 10
4 cells were seeded in 24-well plate containing five-hundred microliter media (without serum) and incubated under aforementioned conditions. Then old media was discarded and five-hundred microliter new media containing 25, 50, 100, and 200 μg/mL of venom were added and incubated for 24 h in 37 °C. The cells then trypsinized and harvested in fresh 1.5 mL tubes and centrifuged in 1500 rpm for 5 min in 4 °C and washed with PBS (pH 7.4) two times. Two-hundred microliter PBS was added to tubes and the cells were singled by a needle. The slides were covered by normal melting 1% (v/v) agarose and incubated for 10 min at 4 °C. The cell suspensions were mixed with low melting 1% (v/v) agarose (1 to 2 ratios) and were added to the slides. To form one cell layer, a glass lamel was applied to every slide. In order to cell lysis and nucleus distraction, all slides were incubated for 16-18 h in fresh and cold lysis buffer (NaCl 2.5M, EDTA 100 mM, Tris 10 mM, NaOH 0.2M, Triton X-100 1% and pH 10) at 4 °C. Then, the slides were washed two times with electrophoresis buffer for 20 min and incubated at fresh electrophoresis buffer for 40 min at 4 °C. The electrophoresis was done in 25 V and 300 mA for 45 min at 4 °C. In order to neutralization, the slides were incubated for 10 min in neutralizing buffer (Tris 0.04 M, pH 7.5). Then, the slides were incubated in one-hundred microliter ethidium bromide (20 μg/mL) for 10 min at room temperature. The slides were washed two times (10 min each) with water and analyzed by fluorescent inverted microscope (Nikon, Japan) and the results were statistically analyzed.
Caspase-3/CPP32 activity assay
To determine the caspase 3 activity in MCF-7 and Vero cells treated with Hottentotta schach crude venom compared with the untreated control cells, Caspase-3/CPP32 Colorimetric Assay Kit (Catalog #K106-100) was used. The assay is based on spectrophotometric detection of the chromophore p-nitroaniline (pNA) after cleavage from the labeled substrate DEVD-pNA. The pNA light emission was quantified using a microtiter plate reader at 400 nm. Comparison of the absorbance of pNA from an apoptotic sample with an uninduced control allows determination of the fold increase in CPP32 activity.
Statistical analysis
The results were reported as mean ± SD and the data were analyzed using GraphPad Prism software. The treated cells and controls were analyzed using ANOVA and Tukey tests. The differences were considered to be significant at P < 0.05 (*), P < 0.01 (**), P < 0.001 (***) and P < 0.0001 (****). All the experiments were conducted no less than 3 times.