
Introduction
Epidermal growth factor receptor (EGFR) is a family of receptor tyrosine kinases which includes human epidermal growth factor receptor HER1-4 (1-4). These tyrosine kinases represent pivotal roles in different stages of epithelial malignancies such as tumor growth, proliferation, and progression (5-9). HER1 and HER2 have been targeted by approved medicine in patients diagnosed with non-small-cell lung cancer (NSCLC) and breast cancer (10, 11). The HER family members are transmembrane proteins and possess an intracellular kinase domain that can participate in cellular signaling after homodimerization through phosphorylation (12). On the contrary, HER3 shows no intrinsic tyrosine kinase properties unless it goes under hetrodimerization and forms dimers such as HER2-HER3 and HER1 (EGFR)-HER3. Having that said, HER3 is called “pseudokinase”. The lack of HER3 kinase activity directly results from mutations at the conserved residues Asp813 and Glu738. No data explains the exact role of HER3 in cancer; however its overexpression in multiple cancer types suggested HER3 as a potential target in cancer therapy (13-15). It is worth noting that different normal human tissues ranging from intestines to respiratory tracts also express this receptor. A wide range of malignancies including prostate, gastric, breast, pancreatic, lung, and ovarian cancers display overexpression of HER3 (Figure 1) (16-19). To that end, HER3-targeted cancer therapy could be an alternative to the anti-EGFR and anti-HER2 resistance therapies. Different agents such as monoclonal antibody seribantumab were investigated for cancer HER3-targeted therapy (20, 21).
Targeting HER3 also can be useful for diagnostic purposes. Different types of HER3 ligands such as short peptides, nanobodies, antibodies, antibody fragments, and affibody molecules were radiolabel and investigated for imaging purposes. The main challenge that restrains the development of HER3-imaging agents is the HER3 normal expression in several adult tissues, including the liver and gastrointestinal tracts (22, 23). Another challenge to developing an imaging radiotracer is the moderate expression of this receptor malignant tissues which is less than 5 × 104 receptors/cell (24, 25). The moderate expression of this receptor in cancer tissues severely reduces the imaging contrast against the background due to the high uptake in normal tissues. The main goal of this review is to focus on the different radiotracers that were investigated for HER3-targeted imaging (Figure 1). Therefore, different aspects of HER3 radiotracers and their advantages and disadvantages will be discussed with copious details.
An insight into HER3 functions and structure both in normal and malignant conditions
Like other EGFR family members, HER3 also possesses three domains, including an extracellular, a transmembrane, and an intracellular domain. The extracellular domain has four different subdomains (I-IV). The subdomains I, II, and III are involved in the ligand-binding process and it is reported that their structure is leucine-rich. On the other hand, subdomains II and IV are crucial for the receptor conformation and stability since they are cysteine-rich and can form disulfide bonds. Moreover, subunit II has a section known as the dimerization loop (26, 27). The intracellular domain of HER3 possesses different subunits, including a juxtamembrane segment, a kinase domain, and a C-terminal domain (28, 29). As mentioned earlier, subunits I and III are involved in ligand binding and binding the ligands such as neuregulins (heregulin and NRG-2), stimulats conformational change in HER3 structures and exposes the dimerization loop to other EGFR family members (30, 31).
HER3 is expressed in the mesenchyme of the endocardial cushion, and it is closely related to the development of the heart valve. To that end, HER3 null mouse embryos displayed highly underdeveloped heart valves. HER3 involvement in neural differentiation and sympathetic nervous system development is confirmed (32-34). The heterodimer of HER3-HER2 is involved in several pathologic conditions such as resistance to different chemotherapy agents and the promotion of invasion and metastasis. Although there is no solid evidence the HER3 alone is involved in malignancies, it is worth noting that some reports claimed that the overexpression of HER2 boosts the formation of HER3-HER2 heterodimer without any need for ligand binding (neuregulins) and cause a weak but continuous signaling activity (35-37).
Single-photon emission computed tomography (SPECT) radiotracers
99mTc-radiotracers
Several γ-emitting isotopes such as 99mTc (half-life 6 h) and 111In (half-life 67 h) for SPECT have been used to label tumor-targeting molecules for imaging purposes. Affibodies are small (7 kDa) three-helical proteins that can provide a unique tool for radionuclide imaging (38-40). To provide a proper HER3-targeted affibody-based radiotracer, an affibody with subnanomolar affinity is required. Two anti-HER3 affibody molecules (Z08698 and Z08699) have very high affinities around 50 and 21 pM, respectively (41). To that end, a histidyl-glytamyl-histidyl-glytamyl-histidyl-glytamyl-tagged Z08699 ((HE)3-Z08699) affibody molecule was radiolabel with 99mTc(CO)3(H2O)3+. The resulted radiotracer (99mTc(CO)3-(HE)3-Z08699) ability to target HER3 was evaluated in-vitro and in-vivo. This radiotracer displayed a high affinity toward HER3 receptors in LS174T and BT474 cell lines and xenografts (Table 1) (42).
111In-radiotracers
The potential of 111In radiolabeled Z08698 for HER3 imagining was also investigated. In order to provide sufficient chelator for 111In radiolabeling different chelators including NOTA (1,4,7-triazacyclononane-N,N′,N′′-triacetic acid), NODAGA (1-(1,3-carboxypropyl)-4,7-carboxymethyl- 1,4,7-triazacyclononane), DOTA (1,4,7,10-tetraazacyclododecane-1,4,7,10-tetraacetic acid), and DOTAGA (1,4,7,10-tetraazacyclododececane,1-(glutaric acid)−4,7,10-triacetic acid) were coupled to the C-terminus of affibody Z08698. This study reported that all the radiolabeled affibody including 111In-DOTAGA-Z08698, 111In-DOTA-Z08698, and 111In-NOTA-Z08698 displayed a high affinity for HER3 in-vitro and in-vivo. The tumor uptakes of all these radiotracers were the same. However, the accumulation of the negatively charged 111In-DOTAGA-Z08698 in the liver was significantly lower than the others (Table 1) (43).
A histidyl-glytamyl-histidyl-glytamyl-histidyl-glytamyl tagged Z08699 ((HEHEHE)-Z08699 or (HE)3- Z08699) affibody molecule conjugated with DOTAGA ((HE)3-Z08698-DOTAGA) was also radiolabeled with 111In to obtain 111In-(HE)3-Z08698-DOTAGA. This radiotracer possesses a high affinity toward HER3. This study also investigates the effect of non-residualizing [125I]-N-succinimidyl-4-iodobenzoate (125I-PIB) on (HE)3-Z08698-DOTAGA (125I-PIB-(HE)3-Z08698-DOTAGA) tumor uptake in comparison with 111In-(HE)3-Z08698-DOTAGA. Both of these radiotracers exhibited a high affinity toward HER3 expressing BxPC-3 and DU145 cancer cell lines. 125I-PIB-(HE)3-Z08698-DOTAGA showed faster clearance than 111In-(HE)3-Z08698-DOTAGA from most tissues, while its blood concentration was significantly higher. These radiotracers were tumor-specific, and the tumor uptake of 125I-PIB-(HE)3-Z08698-DOTAGA was lower. The only advantage of using non-residualizing 125I-PIB for affibody radiolabeling was the higher tumor-to-liver ratio (Table 1) (44).
Positron-emission tomography (PET) radiotracers
68Ga-radiotracers
The high affinity of Z08698 toward HER3 encouraged its radiolabeling with a wide range of radionuclides. Due to the advantages of PET such as high resolution, sensitivity, and quantification accuracy in comparison with SPECT (45,46), PET radiotracers of Z08698 were developed. Therefore, a chelator (NOTA) conjugated version of HEHEHE-Z08698 ((HE)3-Z08698) was radiolabeled with 68Ga (68Ga-(HE)3-Z08698-NOTA). This radiotracer showed a high affinity toward HER3 positive cell lines (BT474, BxPC-3) and their xenografts, while it displayed significantly lower affinity toward A431 cell line with low HER3 expression. The xenograft models of BT474, and BxPC-3 showed high tumor-to-blood and tumor-to-muscle ratios (>20 and >15, respectively) at 3 hours after injection of 68Ga-(HE)3-Z08698-NOTA. Therefore, the combination of PET radionuclide, high affinity toward HER3, and high tumor uptake made 68Ga-HEHEHE-Z08698-NOTA an eligible radiotracer for HER3 targeted tumor imaging (47). In another study, to reduce liver uptake of 68Ga-(HE)3-Z08698-NOTA, the NOTA chelator was replaced with DOTAGA. The negative charge of DOTAGA when it forms a complex with 68Ga could lead to lower hepatic uptake of 68Ga-(HE)3-Z08698-DOTAGA and better imaging properties in comparison with 68Ga-(HE)3-Z08698-NOTA. The hepatic uptake of 68Ga-(HE)3-Z08698-DOTAGA was lower compared with 68Ga-(HE)3-Z08698-NOTA while its blood concentration was higher; hence the quality of the imaging stayed almost the same (Table 1) (48).
In addition to 68Ga-affibodies, 68Ga-peptides are also investigated for their ability to target HER3. HER3P1 is a peptide (the sequence is CLPTKFRSC) and targets the extracellular domain of HER3. The 68Ga labeled HER3P1 (68Ga-NOTA-HER3P1) showed moderate affinity (270 ± 151 nM) toward HER3. 68Ga-NOTA-HER3P1 displayed affinity toward MDA-MB-453 (moderate expression of HER3) which can be blocked using excess unlabeled NOTA-HER3P1. It is worth noting that HER3P1 can distinguish HER3 from EGFR and HER2. 68Ga-NOTA-HER3P1 also showed high accumulation in HER3 high expressing cell line (tumor to muscle was about 1.8) 22RV1 xenograft while its accumulation in HCC-1954 (low expression of HER3) xenograft was much lower (Table 1) (49).
55Co-radiotracers
As it was mentioned earlier, (HE)3-Z08698 was radiolabeled with 68Ga using different chelators. However, the short half-life of 68Ga makes the delayed imaging almost impossible. Therefore, other PET radionuclides such as 55Co with a longer half-life (17.5 h) seem to be a viable option for radiolabeling (HE)3-Z08698. A study reported conjugation of different chelators such as NOTA, NODAGA, DOTA, and DOTAGA to this affibody. The resulted affibodies were radiolabeled with 57Co (271.8 days) as a surrogate for 55Co. Using different conjugated chelator results in a range of metal complexes with various charges and different RCP (radiochemical purity). All the (HE)3-Z08698 displayed high radiochemical yields (>99%) except for 57Co-(HE)3-ZHER3:08698-NOTA which was subject to NAP5 size-exclusion chromatography to improve purity (>99%). 57Co-(HE)3-ZHER3:08698-DOTA and the other conjugates specifically bind to the BxPC-3 and DU145 (HER3-expressing cell lines) which can be blocked by the addition of the cold affibody. All the conjugates display high and almost the same affinity (in subnanomolar range) toward HER3. However, 57Co-(HE)3-ZHER3:08698-DOTA showed the highest tumor-to- muscle ratio and the best imaging contrast at 3 and 24 h post-injection. It is noteworthy, that the application of 57Co as a surrogate of 55Co is more convenient due to its availability and longer half-life. However, the different radionuclide impurities of 55Co and 57Co may lead to different results for 55Co-(HE)3-ZHER3:08698-DOTA in comparison with 57Co-(HE)3-ZHER3:08698-DOTA (Table 1) (50). One of the drawback of 57Co-(HE)3-ZHER3:08698-NOTA is its high non-specific liver uptake so that co-injection of trimer affibody ((ZHER3:08698)3) with three times more molar excesses than 57Co-(HE)3-ZHER3:08698-NOTA concentration has been suggested to significantly reduce hepatic uptake with no impact on its tumor uptake (43).
18F-radiotracers
Radiolabeling of a biomolecule with 18F usually requires a prosthetic group (51, 52). However, there are other approaches, such as using [18F]AlF and a chelator (NOTA) (53, 54). Sometimes a combination of these approaches can be applied. To that end, a study reported the radiolabeling of ZHER3:8698 affibody with 18F using [18F]AlF and a chelator (NOTA). In this study, two different methods were applied to conjugate a chelator to ZHER3:8698 affibody. The first approach introduced a NOTA chelator to the affibody through its cysteine residue using a maleimide functionalized NOTA (NOTA-ZHER3:8698) and then radiolabeled the resulted radiotracer using [18F]AlF to obtain [18F]AlF-NOTA-ZHER3:869. The second approach in this study was radiolabeling of tetrazine functionalized 1,4,7- triazacyclononane-1,4-diacetate (NODA) using [18F]AlF as the source of 18F to obtain a prosthetic group. The resulted molecule was then conjugated to trans-cyclooctene (TCO) functionalized ZHER3:8698 to synthesized [18F]AlF-NODA-ZHER3:8698. Both of the resulted radiotracers showed a high affinity toward HER3. The study indicated that these radiotracers bind to MCF-7 (high-expressing HER3 cell line), which was blocked by an excess amount of the cold affibody. Furthermore, [18F]AlF-NOTA-ZHER3:869 and [18F]AlF-NODA-ZHER3:8698 displayed very low binding to MDA-MB-231 (low expressing HER3 cell line). This study indicated that these radiotracers successfully visualized tumors in the MCF-7 xenograft model (Table 1) (55). To our personal opinion and despite the high affinity of these radiotracers toward HER3, both of these radiotracers need extensive purification after radiolabeling which is not desirable for 18F regarding its 110 minutes’ half-life. Besides, both of these radiotracers displayed high abdomen cavity radioactivity and high blood to tumor ratio (24.61 and 9.3 for 1 h post-injection) which jeopardizes the potential clinical application of these radiotracers. In contrast to 89Zr (t1/2 = 78.41 h), in the case of 18F (t1/2 = 109.7 min) delayed imaging cannot resolve this problem due to the much lower half-life of 18F in comparison with 89Zr.
89Zr-radiotracers
Radiolabeled anti-human HER3 monoclonal antibodies such as Mab#58 are also potential candidates for HER3 imaging. 89Zr-labeled Mab#58 was investigated as a radiotracer for imaging of HER3. Usually, radiolabeling of biomolecules requires the conjugation of a suitable bifunctional chelating agent (BFCA). Mab#58 is no exception; before radiolabeling, it was conjugated with p-isothiocyanatobenzyl-desferrioxamine B as a BFCA for 89Zr. The 89Zr-labeled Mab#58 (89Zr-Mab#58) displayed a high affinity for the RH7777 cell line (HER3 overexpressing cell line), and the calculated Kd was 2.7 nM. The biodistribution of this radiotracer showed significant tumor uptake compared to the control group. The radiotracer accumulation in the tumor tends to increase from day 1 to day 4 post-injection. This radiotracer successfully visualized the tumor in the xenograft model (Table 1) (56). However, we suspect the tumor visualization is mainly due to the radioactivity of the tumor blood content rather than tumor uptake.
GSK2849330 is an anti HER3 monoclonal antibody. To provide sufficient chelating sites for 89Zr-radiolabeling p-isothiocyanatobenzyl-desferrioxamine was conjugated to this biomolecule. The final radioactive biomolecule (89Zr-GSK2849330) can visualize CHL-1 (HER3 expressing cell line) xenograft in mice while it does not show any accumulation in MIA-PaCa-2 (HER3 negative cell line) xenograft. This study suggests that the tumor uptake of 89Zr-GSK2849330 is dose-dependent, and pretreatment of CHL-1 xenograft with GSK2849330 (50 mg/kg) reduces significantly reduces tumor uptake. Confocal microscopy images also confirmed the affinity of fluorescently labeled GSK2849330 toward CHL-1 tumors. Moreover, this study expressed that GSK2849330 can inhibit tumor growth. It should be noticed that this study did not report any quantities in-vitro data (such as Kd, Ki, or IC50) about the affinity of 89Zr-GSK2849330 for HER3 (Table 1) (57). Another study reported the application of 89Zr-GSK2849330 in six patients diagnosed with HER3-positive tumors who were not responsive to standard treatments. The results of this study indicated that 89Zr-GSK2849330 displayed good tumor uptake in all six patients which their HER3 receptors were blocked with pre-treated GSK2849330. Moreover, this study reported an inhibitory dose for 50 and 90 percent (ID50 and ID90) of the patient’s population which were 2 and 18 mg/kg, respectively. However, reporting ID50 and ID90 in a population of six patients is not valid enough (58).
A glycoengineered humanized monoclonal antibody, lumretuzumab (RG7116, RO5479599), can bind to the extracellular domain of HER3; hence it can inhibit its heterodimerization and downstream signaling pathway (59). Furthermore, a phase I clinical study in patients diagnosed via HER3-positive solid tumors indicated that they could tolerate lumretuzumab monotherapy (60). Due to this evidence, 89Zr-radiolabeled lumretuzumab (89Zr-lumretuzumab) has undergone clinical trial. 89Zr-lumretuzumab successfully visualized tumor lesions in patients with locally advanced or metastatic HER3-positive solid tumors. Due to the high uptake of 89Zr-lumretuzumab in the liver, this radiotracer failed to visualize tumor lesions in the liver while these lesions were visible on the computed tomography (CT) scan (61).
Regarding the fact that HER3-mediated resistance occurs when using Hsp90 inhibitors such as AUY922 (62, 63), ZHER3:8698 affibody molecule was radiolabeled with 89Zr not only as an imaging radiotracer but also as a tool for evaluation of HER3 expression levels in breast cancer xenograft models. To that end, first ZHER3:8698 was conjugated to deferoxamine-maleimide and then radiolabeled with 89Zr to obtain 89Zr-DFO-ZHER3:8698. The study suggests that 89Zr-DFO-ZHER3:8698 can visualize the tumor in MCF-7 xenograft. The treatment of the xenograft model with AUY922 (Hsp90 inhibitors) resulted in therapy resistance through HER3 up-regulation. To that end, the uptake of 89Zr-DFO-ZHER3:8698 in MCF-7 tumor was increased by 1.51 fold from day 0 (no AUY922) to 14 days after treatment with AUY922 (Table 1) (64). A similar study applied the 89Zr radiolabeled monoclonal antibody (89Zr-mAb3481) to evaluate the effect of lapatinib treatment on HER3 expression and the internalization of mAb3481 monoclonal antibody (65).
MEHD7945A (duligotuzumab) is a fully human IgG1 mAb that has affinity toward EGFR (KD ~ 1.9 nM) and HER3 (KD ~ 0.4 nM) (66,67). MEHD7945A was developed as an alternative for treating solid tumors that are resistant to EGFR-targeted treatment through HER3-mediated pathways (68,69). MEHD7945A bold characteristic in the treatment of locally advanced or metastatic epithelial cancers is its very low toxicity (70). To that end, MEHD7945A was radiolabeled with 89Zr (89Zr-MEHD7945A) for spontaneous imaging of EGFR and HER3. For in-vitro study of 89Zr-MEHD7945A three different pancreatic cell lines (Mia PACA2 (negative control), AsPC-1 and BxPC-3) with EGFR and HER3 expression were selected. The order of the HER3 expression in these cell lines is AsPC-1> BxPC-3> Mia PACA2. The uptake of 89Zr-MEHD7945A in these cell lines was time-dependent, and the affinity of 89Zr-MEHD7945A toward AsPC-1 and BxPC-3 was in the same range. This study reported that blockage of EGFR with cetuximab in AsPC-1 xenograft tumors decreased the tumor uptake of the radiotracer up to three folds while blocking the HER3 receptors with DL3.6b did not alter the radiotracer uptake. BxPC-3 xenografts blocking with cetuximab did not affect the radiotracer uptake, while the study indicated that blocking with DL3.6b lead to increasing the radiotracer uptake. In the end, the author of this study suggested that the expression of HER3 in comparison with EGFR is five-fold lower (using flow cytometry data) in both AsPC-1 and BxPC-3. Therefore, the combination of marginal changes in DL3.6b blocked HER3, and a higher level of EGFR expression may put the changes below the sensitivity threshold of 89Zr-MEHD7945A (Table 1) (71). In our personal view, it seems that 89Zr-MEHD7945A is an EGFR imaging probe rather than HER3.
MSB0010853 is a nanobody that is capable of binding to two different epitopes of HER3. Therefore, it can be a robust platform for the development of a HER3 targeted radiotracer. To that end, a study reported 89Zr radiolabeled MSB0010853 (89Zr-MSB0010853) for PET imaging of HER3 receptors. 89Zr-MSB0010853 successfully recognized the HER3 receptors of the H441 xenografted model in a dose-dependent manner which was blocked using an excess amount of cold MSB0010853. This study indicated that after 24 hours (post-injection) the accumulation of the radiotracer in the tumor reached its maximum and the tumor site in H441 and FaDu (HER3 positive cell line) xenografted was visualized while the tumor site in Calu-1 (negative HER3 cell line) is not observable (Table 1) (72). It should be noticed that the MSB0010853 nanobody contains a binding site for albumin and can cross-reactive with albumin and HER3. Moreover, the tumor and blood radioactivity uptakes at 24 h post-injection are very close together. Therefore, to our personal opinion, it is not clear how much of tumor uptake is related to the interaction of HER3 and 89Zr-MSB0010853 and how much is related to tumor blood content.
11C- radiotracers
AZD8931 ((73)) is a known small molecule with a tyrosine kinase inhibitory effect. This molecule possesses equipotent affinity toward EGFR, HER2, and HER3 (74, 75). Therefore, it was radiolabeled with 11C, ([11C]AZD8931), to image HER3 receptors. The high affinity of [11C]AZD8931 toward HER3 and high specific activity made it a potential radiotracer for PET imaging of HER3 (Table 1) (73).
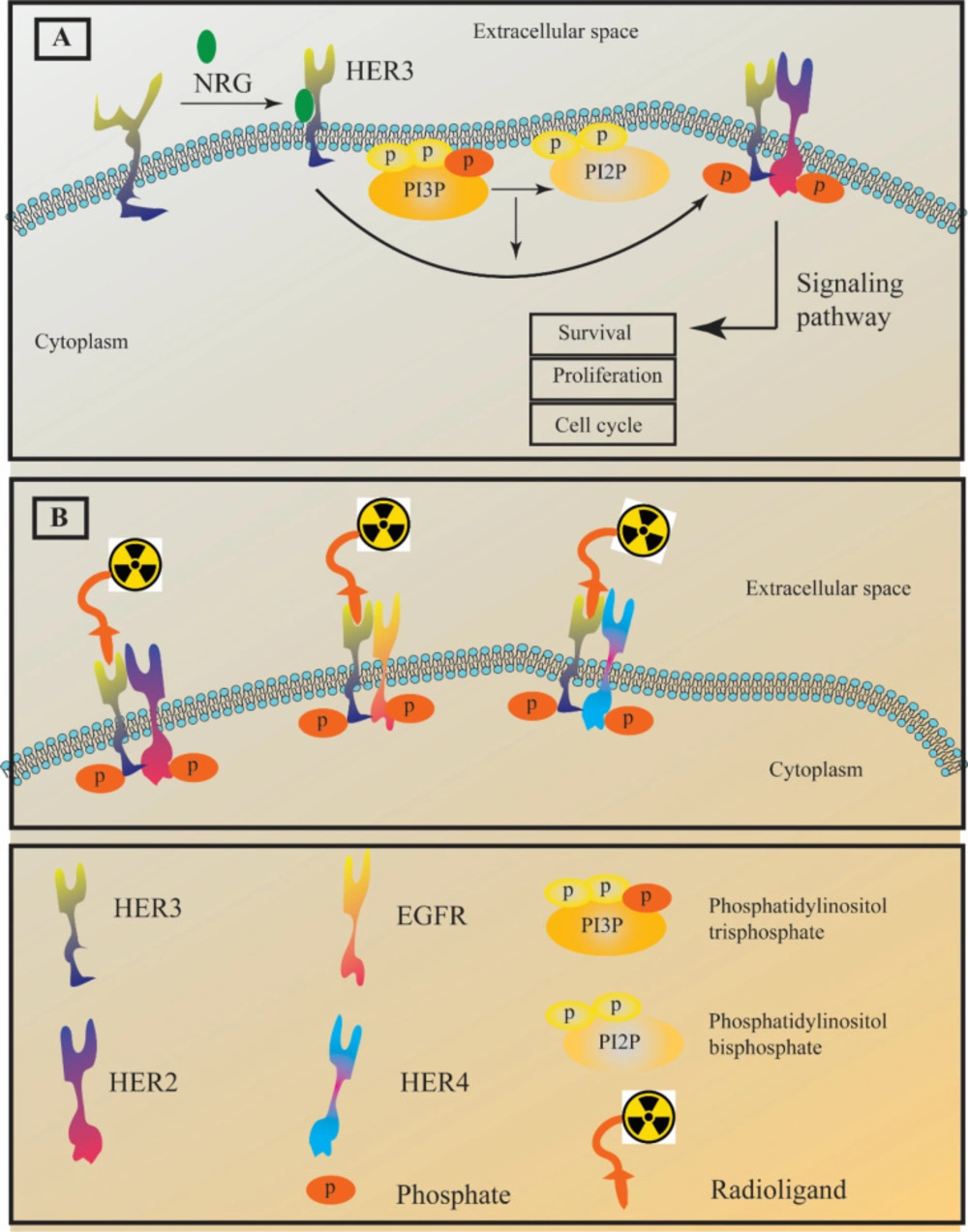
Figure 1
(A) The HER3 receptor adapts an active form through interaction with a ligand known as neuregulin (NRG). HER3 tyrosine kinase domain is impaired; hence, it only undergoes a weak auto-phosphorylation process. The scheme shows the hetrodimerization of HER3/HER2. However, in a similar manner it can form heterodimers with EGFR and HER4. (B) The concept behind HER3 imaging is the attachment of a radioligand to HER3 heterodimers
Table 1
A list of HER3 radiotracers and their characteristics
| No. | Radiotracer | RCP (%) | Type of molecule | Imaging modality | Affinity | Specific activity | Cell line | Tumor to muscle ratio | Tumor to blood ratio | Tumor to liver ratio | Ref. |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 2 | 99mTc(CO)3-(HE)3-Z08699 | >99 | Affibody | SPECT | KD = 27 pM | 1.6 MBq/μg | LS174T and BT474 | 11-12 (4 h p.i.)a | 6-7 (4 h p.i.) | <0.5 (4 h p.i.) | (42) |
| 3 | 111In-DOTAGA-Z08698 | >99 | Affibody | SPECT | KD = 8 ± 6 pM | - | BxPC-3 and DU145 | 21 (24 h p.i.) | 6 (4 h p.i.) | 0.61 (4 h p.i.) | (43) |
| 4 | 111In-(HE)3-Z08698-DOTAGA | >99 | Affibody | SPECT | KD = 19 ± 1 pM | 1.2 MBq/μg | BxPC-3 and DU145 | 1.63 (4 h p.i.) | 43 (4 h p.i.) | 0.7 (4 h p.i.) | (44) |
| 5 | 68Ga-(HE)3-Z08698-NOTA | >98 | Affibody | PET | - | 16–19 GBq/μmol | LS174T, BxPC-3, BT474 | >15 (3 h p.i.) | 2 (1 h p.i.) | 0.95 (1 h p.i.) | (47) |
| 6 | 68Ga-NOTA-HER3P1 | >95 | Peptide | PET | KD = 270 ± 151 nM | 296 ± 25.9 MBq/mg | 22RV1 | 1.8 (1 h p.i.) | 2.78 (1 h p.i.) | 0.71 (1 h p.i.) | (49) |
| 7 | 57Co-(HE)3-ZHER3:08698-DOTA | 99.7 | Affibody | SPECT | KD = 0.4 ± 0.8 nM | - | BxPC-3 and DU145 | 28±4 (24 h p.i.) | 18 (24 h p.i.) | 1.6 (24 h p.i.) | (50) |
| 8 | [18F]AlF-NOTA-ZHER3:869 | >98 | Affibody | PET | KD = 0.44 ± 0.04 nM | 0.8-1.5 MBq | MCF-7 | 34.96 (1 h p.i.) | 24.61 (1 h p.i.) | 0.77 (1 h p.i.) | (55) |
| 9 | [18F]AlF-NODA-ZHER3:8698 | >95 | Affibody | PET | KD = 1.01 ± 0.28 nM | 0.7-2.3 MBq/μg | MCF-7 | 18.01(1 h p.i.) | 9.27 (1 h p.i.) | 0.86 (1 h p.i.) | (55) |
| 10 | 89Zr-Mab#58 | >90 | Monoclonal antibody | PET | KD = 2.7 nM | 40–110 kBq/μg | RH7777 | >10 (4 days p.i.) | 1.1 (4 days p.i.) | 1.3 (4 days p.i.) | (56) |
| 11 | 89Zr-DFO-ZHER3:8698 | >95 | Affibody | PET | KD = 0.55 ± 0.05 nM | 2.5 - 2.7 MBq/μg | MCF-7 | 20.4 (3 h p.i.) | 4.75 (3 h p.i.) | 1.2 (3 h p.i.) | (64) |
| 12 | 89Zr-MEHD7945A | >95 | Monoclonal antibody | PET | IC50 = 0.37-0.48 nM | 25.5 ± 3.7 MBq/nmol | BxPC-3 and AsPC-1 | >20 (96 h p.i.) | >6 (96 h p.i.) | >5 (96 h p.i.) | (71) |
| 13 | 89Zr-MSB0010853 | >95 | Nanobody | PET | - | - | H441 and FaDu | >10 (24 h p.i.) | 8 (24 h p.i.) | <1 (24 h p.i.) | (72) |
| 14 | [11C]AZD8931 | >99 | Small molecule | PET | IC50 = 4 nM | 370–1110 GBq/µmol | - | - | - | - | (73) |
Conclusion
The exact role of the HER3 receptor in malignancies is still unclear. However, this receptor is certainly linked to therapy resistance and poor prognosis of some cancers; hence, trying to develop a clinically useful radiotracer for imaging of this radiotracer seems reasonable. Due to a few difference between HER3 and other members of the human epidermal growth factor receptor family, such as HER2 developing radiotracer for HER3 imaging is much more challenging because: firstly; even in the case of HER3 overexpression in cancer, the number of HER3 does not exceed 50000 receptors/cell and, secondly; normal human tissues, such as the respiratory, urinary, gastrointestinal, and reproductive tract and even skin physiologically express HER3 receptor. To that end, to develop a radiotracer for HER3, the affinity of the tracer should be very high toward HER3 (preferably in the picomolar range), such as different radiotracer of ZHER3:08698 affibody. Furthermore, such radiotracers are more effective when they are radiolabel with longer half-life radionuclide. To that end, the 89Zr (t1/2 = 78.4 h) radiotracers are much more desirable and practical in comparison with 18F (t1/2 = 109.7 min) radiotracer, due to the fact that delayed imaging with 89Zr radiotracer is possible.
Conflict of interest
The authors declare no conflict of interest.
