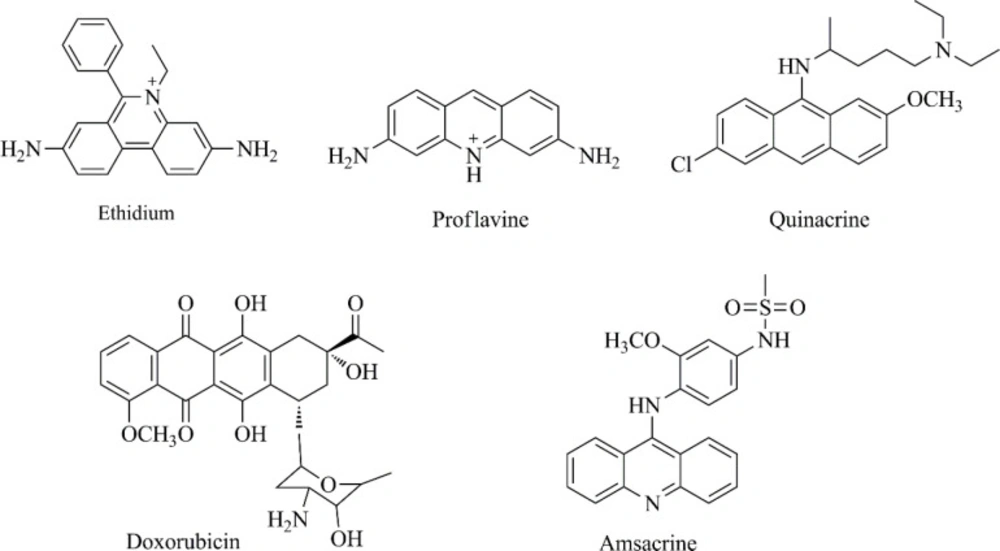
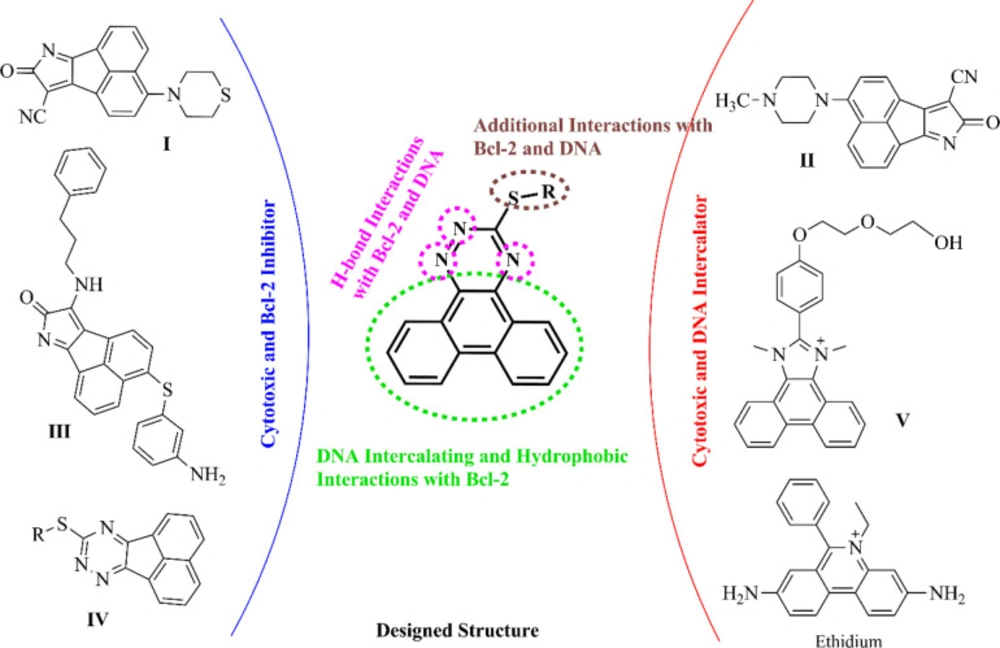
Chemistry
Apparatus and reagents
Melting points were measured on a hot stage apparatus (Electrothermal, Essex, UK) and were uncorrected. 1H spectra were recorded on Brucker 500 spectrometer, using TMS as an internal standard, and Chemical shifts (δ) are reported as ppm. IR spectra were obtained on a Perkin-Elmer spectrometer (KBr disk) (Perkin-Elmer, Waltham, MA). Mass spectra were recorded with an Agilent spectrometer (Agilent technologies 9575c inert MSD, USA). All commercially available solvents and reagents were of analytical grade and obtained from Merck and Aldrich.
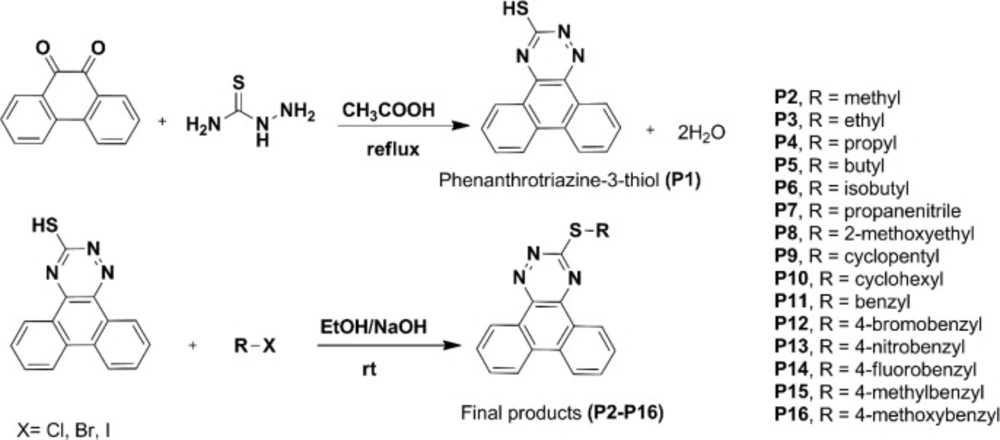
Procedure for the synthesis of phenanthro [9,10-e][1,2,4]triazine-3-thiol (P1)
The phenanthrene-9,10-dione (4.16 g, 20 mmol) was added to 80 mL of acetic acid, and the mixture was heated to about 100 oC with stirring for about half an hour. Thiosemicarbazide (3.65 g, 40 mmol) was added, and the mixture was refluxed for about three hours. The reaction mixture cooled to room temperature and then poured into ice-cold water. The solid product was filtered and washed with cold water. Yield: 67.5%, Rf = 0.56 (EtOAc/ PetEther 1:1), mp: 122-124 °C. 1H NMR (500 M HZ, CDCl3): δH 14.65 ppm (s, 1H, NH), 8.74-8.39 (m, 2H, phenanthrene-H), 8.19-8.06 (m, 5H, phenanthrene-H), 7.78-7.37 (m, 9H, phenanthrene-H), 6.72 (s, 1H, SH). MS (EI) m/z (%) 263 (38.6), 235 (77.2), 176.1 (100), 150 (10.53). IR (KBr): υ = 3145 (CH-aromatic), 3413 (NH), 3242 cm-1 (SH).
General procedure for the synthesis of phenanthro[9,10-e][1,2,4]triazine-3-thiol derivatives (P2-P16)
The phenanthro[9,10-e][1,2,4]triazine-3-thiol (0.263 g, 1 mmol) was added to a stirred hot ethanolic solution of sodium hydroxide (0.1 M). After cooling to room temperature, corresponding alkyl/benzyl halide was added, and the mixture was stirred for about 2-24 h at 25 °C. Completion of the reaction was checked by thin-layer chromatography (TLC). The solvent was reduced under vacuum; the precipitate was filtered and washed with ethanol and cold water. Finally, the dried precipitate was purified by recrystallization or chromatography to give the final pure derivative (
Table 1).
3-(methylthio)phenanthro[9,10-e][1,2,4]tri-azine (P2)
Compound P2 was prepared by the described procedure using methyl iodide (3 mmol, 0.19 mL) and the obtained precipitate was recrystallized from ethyl acetate. Yield: 36.1%, Rf = 0.67 (EtOAc/ PetEther 1:1), mp: 157-160 °C. 1H NMR (500 M HZ, CDCl3): δH 9.36 (d, 1H, J = 7.2 Hz, phenanthrene-H), 9.15 (d, 1H, J = 7.6 Hz, phenanthrene-H), 8.53 (s, 2H, phenanthrene-H), 7.88-7.72 (m, 4H, phenanthrene-H), 2.89 ppm (s, 3H, CH3). MS (EI) m/z (%) 277.1 (43.9), 249.2 (79), 235 (46.5), 203.1 (7), 176.1 (100), 150.1 (14). IR (KBr): υ = 3060 (CH-aromatic), 2927 cm-1 (CH-aliphatic).
3-(ethylthio)phenanthro[9,10-e][1,2,4]tri-azine (P3)
Compound P3 was prepared by the described procedure using ethyl iodide (3 mmol, 0.24 mL) and the obtained precipitate was recrystallized from ethyl acetate. Yield: 54.91%, Rf = 0.67 (EtOAc/ PetEther 1:1), mp: 131-133 °C. 1H NMR (500 M HZ, CDCl3): δH 9.4 (d, 1H, J = 7.5 Hz, phenanthrene-H), 9.2 (d, 1H, J = 7.5 Hz, phenanthrene-H), 8.6 (t, 2H, J = 8 Hz, phenanthrene-H), 7.92-7.74 (m, 4H, phenanthrene-H), 3.5 (q, 2H, J = 7.2 Hz, CH2-CH3), 1.6 ppm (t, 3H, J = 7.2 Hz, CH2-CH3). MS (EI) m/z (%) 291.1 (40.35), 263.1 (28), 235.1 (100), 203.1 (10), 176.1 (75.44), 150.1 (10.5). IR (KBr): υ = 3065 (CH-aromatic), 2928 cm-1 (CH-aliphatic).
3-(propylthio)phenanthro[9,10-e][1,2,4]tri-azine (P4)
Compound P4 was prepared by the described procedure using 1-bromopropane (3 mmol, 0.29 mL), the obtained precipitate was dissolved in appropriate solvent and purified using silica gel column chromatography with petroleum ether-ethyl acetate as the mobile phase. Yield: 49.12%, Rf = 0.69 (EtOAc/ PetEther 3:7), mp: 118-120 °C. 1H NMR (500 MHZ, CDCl3): δH 9.37 (d, 1H, J = 7.0 HZ, phenanthrene-H), 9.15 (d, 1H, J = 7.6 HZ, phenanthrene-H), 8.55 (t, 2H, J = 7.9 HZ, phenanthrene-H), 7.90-7.30 (m, 4H, phenanthrene-H), 3.48 (t, 2H, J = 7.2 HZ, CH2-CH2-CH3), 2.0 (sext, 2H, -CH2-CH2-CH3), 1.22 ppm (t, 3H, J = 7.3 HZ, -CH2-CH2-CH3). MS (EI) m/z (%) 305.1 (28), 279.1 (36.9), 263.1 (47.4), 235.1 (100), 203.1 (14), 176.1 (77.2), 150.1 (14). IR (KBr): υ = 3071 (CH-aromatic), 2960, 2929 cm-1 (CH-aliphatic).
3-(butylthio)phenanthro[9,10-e][1,2,4]tri-azine (P5)
Compound P5 was prepared by the described procedure using 1-bromobutane (3 mmol, 0.29 mL), the obtained precipitate was purified using silica gel column chromatography with petroleum ether-ethyl acetate as the mobile phase and then recrystallized from chloroform. Yield: 14%, Rf = 0.66 (EtOAc/ PetEther 3:7), mp: 81-83 °C. 1H NMR (500 MHZ, CDCl3): δH 9.31 (d, 1H, J = 7.4 HZ, phenanthrene-H), 9.06 (d, 1H, J = 7.4 HZ, phenanthrene-H), 8.47 (m, 2H, phenanthrene-H), 7.85-7.67 (m, 4H, phenanthrene-H), 3.48 (t, 2H, J = 7.3 HZ, CH2-(CH2)2-CH3), 1.96 (pent, 2H, CH2-CH2-CH2-CH3), 1.66 (sext, 2H, CH2-CH2-CH2-CH3), 1.07 ppm (t, 3H, J = 7.3 HZ, CH2-CH2-CH2-CH3). MS (EI) m/z (%) 319.2 (50.9), 291.1 (19.3), 263.1 (52.6), 235.1 (100), 203.1 (64.9), 176.1 (75.4), 150.1 (14). IR (KBr): υ = 3071 (CH-aromatic), 2963 cm-1 (CH-aliphatic).
3-(isobutylthio)phenanthro[9,10-e][1,2,4]tri-azine (P6)
Compound P6 was prepared by the described procedure using 1-bromopropane2-methylpropan (3 mmol, 0.33 ml), the obtained precipitate was purified using silica gel column chromatography with petroleum ether-ethyl acetate as the mobile phase and then recrystallized from chloroform. Yield: 17.8%, Rf = 0.67 (EtOAc/ PetEther 2:8), mp: 93-96 °C. 1H NMR (500 MHZ, CDCl3): δH 9.4 (d, 1H, J = 7.9 HZ, phenanthrene-H), 9.2 (d, 1H, J = 7.9 Hz, phenanthrene-H), 8.6 (t, 2H, J = 9 HZ, phenanthrene-H), 7.93-7.75 (m, 4H, phenanthrene-H), 3.4 (d, 2H, J = 6.8 HZ, CH2(CH(CH3)2)), 2.3-2.1 (m , 1H, CH2(CH(CH3)2)), 1.23 ppm (d, 6H, J = 6.8 Hz, (CH3)2). MS (EI) m/z (%) 319.2 (10.5), 291.1 (17.5), 263.1 (75.4), 235.1 (100), 203.1 (10.5), 176.1 (49), 150.1 (8.8). IR (KBr): υ = 3060 (CH-aromatic), 2963, 2926 cm-1 (CH-aliphatic).
3-(phenanthro[9,10-e][1,2,4]triazin-3-ylthio)propanenitrile (P7)
Compound P7 was prepared by the described procedure using 3-bromopropionitrile (3 mmol, 0.25 mL) and the obtained precipitate was recrystallized from ethyl acetate. Yield: 25.3%, Rf = 0.27 (EtOAc/ PetEther 3:7), mp: 128-130 °C. 1H NMR (500 MHZ, CDCl3): δH 9.4 (d, 1H, J = 7.8 HZ, phenanthrene-H), 9.17 (d, 1H, J = 7.8 HZ, phenanthrene-H), 8.62 (t, 2H, J = 8.6 HZ, phenanthrene-H), 7.93-7.75 (m, 4H, phenanthrene-H), 3.76 (t , 2H, J = 7.2 HZ, CH2-CH2-CN), 3.15 ppm (t, 2H, J = 7.2 HZ, CH2-CH2-CN). MS (EI) m/z (%) 316.2 (33.3), 288.1 (100), 260.1 (17.5), 235.1 (21.9), 201.1 (4.4), 176.1 (73.7), 150.1 (12.3). IR (KBr): υ = 3064 (CH-aromatic), 2923 (CH-aliphatic), 2245 cm-1 (CN).
3-((2-methoxyethyl)thio)phenanthro[9,10-e][1,2,4]triazine (P8)
Compound P8 was prepared by the described procedure using 2-methoxyethyl chloride (3 mmol, 0.29 ml), the obtained precipitate was purified using silica gel column chromatography with petroleum ether-ethyl acetate as the mobile phase and then recrystallized from ethyl acetate. Yield: 16.2%, Rf = 0.49 (EtOAc/ PetEther 3:7), mp: 100-102 °C. 1H NMR (500 MHZ, CDCl3): δH 9.25 (d, 1H, J = 7.8 HZ, phenanthrene-H), 9.0 (d, 1H, J = 7.8 HZ, phenanthrene-H), 8.42-8.39 (m, 2H, phenanthrene-H), 7.82-7.63 (m, 4H, phenanthrene-H), 3.91 (t, 2H, J = 6.5 Hz, CH2-CH2-OCH3), 3.69 (t, 2H, J = 6.5 Hz, CH2-CH2-OCH3), 3.5 ppm (s, 3H, OCH3). MS (EI) m/z (%) 321 (10.5), 293 (7), 263 (98.3), 23 (100), 203 (73.7), 176 (63.2), 150 (17.5). IR (KBr): υ = 3065 (CH-aromatic), 2962, 2927 cm-1 (CH-aliphatic).
3-(cyclopentylthio)phenanthro[9,10-e][1,2,4]triazine (P9)
Compound P9 was prepared by the described procedure using cyclopentyl bromide (3 mmol, 0.32 mL), the obtained precipitate was purified using silica gel column chromatography with petroleum ether-ethyl acetate as the mobile phase and then recrystallized from ethyl acetate. Yield: 11.6%, Rf = 0.73 (EtOAc/ PetEther 2:8), mp: 170-172 °C. 1H NMR (500 MHZ, CDCl3): δH 9.34 (d, 1H, J = 7.8 HZ, phenanthrene-H), 9.11 (d, 1H, J = 8 HZ, phenanthrene-H), 8.50 (t, 2H, J = 7.7 HZ, phenanthrene-H), 7.87-7.69 (m, 4H, phenanthrene-H), 4.41 (quint, 1H, Hₐ-cyclopenthyl), 2.51-2.46 (m, 2H, H-cyclopenthyl), 1.95-1.82 ppm (m, 6H, H-cyclopenthyl). MS (EI) m/z (%) 331.1 (29.9), 303.1 (22.8), 279.1 (8.8) 262.1 (29.9), 235. 1(100), 208.1 (91.2), 176.1 (63.2), 149 (52.6). IR (KBr): υ = 3032 (CH-aromatic), 2959 cm-1 (CH-aliphatic).
3-(cyclohexylthio)phenanthro[9,10-e][1,2,4]triazine (P10)
Compound P10 was prepared by the described procedure using cyclohexyl bromide (3 mmol, 0.37 mL), the obtained precipitate was purified using silica gel column chromatography with petroleum ether-ethyl acetate as the mobile phase and then recrystallized from ethyl acetate. Yield: 15.8%, Rf = 0.49 (EtOAc/ PetEther 2:8), mp: 144-147 °C. 1H NMR (300 MHZ, CDCl3): δH 9.27 (d, 1H, J = 6 HZ, phenanthrene-H), 9.05 (d, 1H, J = 6 HZ, phenanthrene-H), 8.45 (t, 2H, J = 6 HZ, phenanthrene-H), 7.78-7.61 (m, 4H, phenanthrene-H), 4.13 (quint, 1H, Hₐ-cyclohexyl), 1.84-1.80 (m, 2H, H-cyclohexyl), 1.65-1.50 ppm (m, 4H, H-cyclohexyl), 1.37-1.18 ppm (m, 4H, H-cyclohexyl). MS (EI) m/z (%) 345.2 (28), 317.2 (14), 264.1 (31.6), 235.1 (100), 203.1 (17.5), 176.1 (42), 150.1 (7). IR (KBr): υ = 3060 (CH-aromatic), 2962, 2924 cm-1 (CH-aliphatic).
3-(benzylthio)phenanthro[9,10-e][1,2,4]tri-azine (P11)
Compound P11 was prepared by the described procedure using benzyl bromide (2 mmol, 0.340 g), the obtained precipitate was purified using silica gel column chromatography with petroleum ether-ethyl acetate as the mobile phase and then recrystallized from ethyl acetate. Yield: 41.4%, Rf = 0.65 (EtOAc/PetEther 1:4), mp: 167-170 °C. 1H NMR (300 M Hz, CDCl3): δH 9.122 (d, 1H, J = 5.7 Hz, phenanthrene-H), 8.887 (d, 1H, J = 7.2 Hz, phenanthrene-H), 8.275-8.245 (m, 2H, Ar-H), 7.655-7.172 (m, 9H, Ar-H), 4.608 ppm (s, 2H, CH2 -Phenyl). MS (EI) m/z (%) 353.1 (58.2), 190.1 (100), 91.1 (61.8). IR (KBr): ν 3025 (CH-aromatic), 2963 cm-1 (CH-aliphatic).
3-((4-bromobenzyl)thio)phenanthro[9,10-e][1,2,4]triazine (P12)
Compound P12 was prepared by the described procedure using 4-bromobenzyl bromide (2 mmol, 0.500 g), the obtained precipitate was purified using silica gel column chromatography with petroleum ether-ethyl acetate as the mobile phase and then recrystallized from ethyl acetate. Yield: 48.8%, Rf = 0.62 (EtOAc/PetEther 1:4), mp: 171-175 °C. 1H NMR (300 M HZ, CDCl3): δH 9.304-9.273 (dd, 1H, J = 7.5, 1.8 Hz, phenanthrene-H), 9.065 (d, 1H, J = 8.1 Hz, phenanthrene-H), 8.508-8.461 (t, 2H, J = 7.2 Hz, Ar-H), 7.841-7.627 (m, 4H, Ar-H), 7.389 (s, 4H, Ar-H), 4.598 ppm (s, 2H, CH2 -Phenyl). MS (EI) m/z (%) 90.1 (18.3), 190.1 (100), 170.9 (57.9), 433.0 (56.2). IR (KBr): ν 3062 (CH-aromatic), 2960 cm-1 (CH-aliphatic).
3-((4-nitrobenzyl)thio)phenanthro[9,10-e][1,2,4]triazine (P13)
Compound P13 was prepared by the described procedure using 4-nitrobenzyl bromide (2 mmol, 0.430 g). The obtained precipitate was purified using silica gel column chromatography with petroleum ether-ethyl acetate as the mobile phase and then recrystallized from ethyl acetate. Yield: 16.6%, Rf = 0.46 (EtOAc/PetEther 1:4), mp: 198-202 °C.1H NMR (300 M HZ, CDCl3): δH 9.356 (d, 1H, J = 7.8 Hz, phenanthrene-H), 9.105 (d, 1H, J = 7.8 Hz, phenanthrene-H), 8.589-8.540 (t, 2H, J = 7.2 Hz, Ar-H), 8.196 (d, 2H, J = 8.4 Hz, Ar-H), 7.896-7.702 (m, 6H, Ar-H), 4.787 ppm (s, 2H, CH2 -Phenyl). MS (EI) m/z (%) 398.1 (44.3), 322.3 (66.9), 190.0 (100). IR (KBr): ν 3076 (CH-aromatic), 2923 (CH-aliphatic), 1524, 1354 (ArNO2), 872 cm-1 (C-NO2).
3-((4-fluorobenzyl)thio)phenanthro[9,10-e][1,2,4]triazine (P14)
Compound P14 was prepared by the described procedure using 4-fluorobenzyl chloride (2 mmol, 0.288 g), the obtained precipitate was purified using silica gel column chromatography with petroleum ether-ethyl acetate as the mobile phase and then recrystallized from ethyl acetate. Yield: 61.2%, Rf = 0.60 (EtOAc/PetEther 1:4), mp: 157-160 °C. 1H NMR (300 M HZ, CDCl3): δH 9.171-9.141 (dd, 1H, J = 7.2, 1.5 Hz, phenanthrene-H), 8.910 (d, 1H, J = 7.8 Hz, phenanthrene-H), 8.326-8.288 (m, 2H, Ar-H), 7.692-7.438 (m, 6H, Ar-H), 6.976-6.919 (m, 2H, Ar-H), 4.581 ppm (s, 2H, CH2 -Phenyl). MS (EI) m/z (%) 371.1 (79.3), 343.1 (35.1), 190.1 (73.9), 109.0 (100) IR (KBr): ν 3070 (CH-aromatic), 2963 cm-1 (CH-aliphatic).
3-((4-methylbenzyl)thio)phenanthro[9,10-e][1,2,4]triazine (P15)
Compound P15 was prepared by the described procedure using 4-methylbenzyl chloride (2 mmol, 0.280 g), the obtained precipitate was purified using silica gel column chromatography with petroleum ether-ethyl acetate as the mobile phase and then recrystallized from ethyl acetate. Yield: 34.7%, Rf = 0.65 (EtOAc/PetEther 1:4), mp: 138-141 °C. 1H NMR (300 M HZ, CDCl3): δH 9.013-8.990 (m, 1H, phenanthrene-H), 8.766-8.724 (t, 1H, J = 7.2 Hz, phenanthrene-H), 8.112-8.085 (m, 2H, Ar-H), 7.574-7.409 (m, 2H, Ar-H), 4.561 & 4.526 (2s, 2H, CH2-Phenyl), 2.227 ppm (s, 3H, CH3-Phenyl). MS (EI) m/z (%) 91.1 (32.1), 105.5 (100), 190.1 (96.2), 367.1 (89.1) IR (KBr): ν 3074 (CH-aromatic), 2926 cm-1 (CH-aliphatic).
3-((4-methoxybenzyl)thio)phenanthro[9,10-e][1,2,4]triazine (P16)
Compound P16 was prepared by the described procedure using 4-methoxybenzyl chloride (2 mmol, 0.312 g), the obtained precipitate was purified using silica gel column chromatography with petroleum ether-ethyl acetate as the mobile phase and then recrystallized from ethyl acetate. Yield: 31.2%, Rf = 0.57 (EtOAc/PetEther 1:4), mp: 152-154 °C. 1H NMR (300 M Hz, CDCl3): δH 9.230-9.199 (dd, 1H, J = 6.9, 1.5 Hz, phenanthrene-H), 9.006-8.975 (dd, 1H, J = 8.1, 1.2 Hz, phenanthrene-H), 8.371-8.334 (m, 2H, Ar-H), 7.777-7.572 (m, 4H, Ar-H), 7.501-7.472 (d, 2H, J = 8.7 Hz, ArH), 6.891-6.862 (d, 2H, J = 8.7 Hz, Ar-H), 4.646 (s, 2H, CH2-Phenyl), 3.777 ppm (s, 2H, O-CH3). MS (EI) m/z (%) 383.1 (44.8), 190.0 (31.1), 121.0 (100). IR (KBr): ν 3068 (CH-aromatic), 2952 (CH-aliphatic), 1249, 1045 cm-1 (C-O-C).
Biological section
Cell lines and cell culture
This study used two cell lines, including MOLT-4 (human acute lymphoblastic leukemia) and MCF-7 (human breast adenocarcinoma). MOLT-4 was grown in suspension, while MCF-7 was grown in monolayer culture. The MOLT-4 cell line was purchased from the National Cell Bank of Iran, Pasteur Institute, Tehran, Iran, while MCF-7 cells were obtained from the Iranian Biological Resource Center (IBRC), Tehran, Iran. The cells were cultured in RPMI 1640 supplemented with 10% fetal bovine serum (FBS) and 100 units/mL penicillin-G and 100 µg/mL streptomycin at 37 °C in humidified air containing 5% CO2. RPMI-1640, FBS, trypsin and phosphate-buffered saline (PBS) were purchased from Biosera (Ringmer, UK). 3-(4,5-Dimethylthiazol- 2-yl)-2,5-diphenyltetrazolium bromide (MTT) was obtained from Sigma-Aldrich (St. Louis, MO, USA). Standard chemotherapeutic agents, dimethylsulfoxide (DMSO) and penicillin/streptomycin were purchased from EBEWE Pharma (Unterach, Netherlands), Merck (Darmstadt, Germany) and Invitrogen (San Diego, CA, USA), respectively.
MTT assay
Cells were seeded into 96-well plates with a density of 5×104 cells/ml (100 µl in each well) and incubated for 24 h at 37 °C. Afterward, the synthesized compounds were added to each well (in triplicates) at three different concentrations. Doxorubicin and cisplatin were used as positive controls. The plates were further incubated for 72 h, and then 80 μL of the medium was removed, and the same volume of an MTT solution was added at a final concentration of 0.5 mg/mL. Plates were incubated for 4 h at 37 °C to allow the formazan crystals to be formed. Then,200 μL DMSO was added to each well to dissolve the formazan crystals. Absorbance was determined at 570 nm with background correction at 655 nm using a Bio-Rad microplate reader (Model 680). The percentage of viability inhibition compared to control was assessed for each concentration of the compound, and IC50 values were computed with CurveExpert software version 1.34 for Windows.
Molecular docking analysis
Computational docking, an extremely useful tool to achieve deep insight into drug-target interactions, has played a crucial role in drug discovery. The synthesized derivatives were subjected to molecular docking studies using AutoDock Tools and AutoDock software in this study.
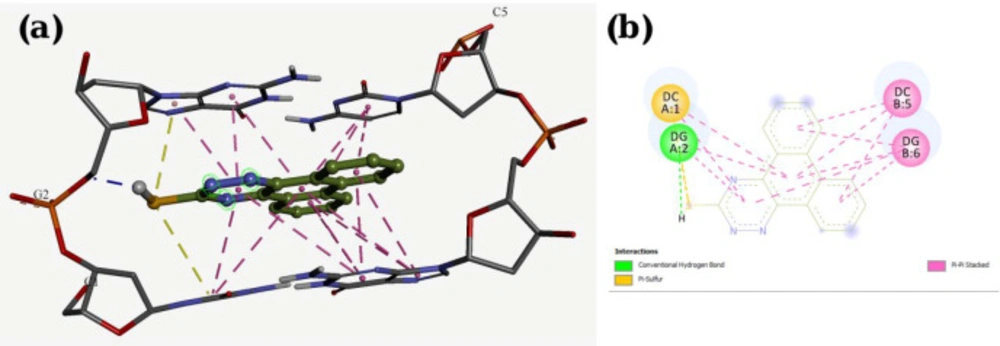
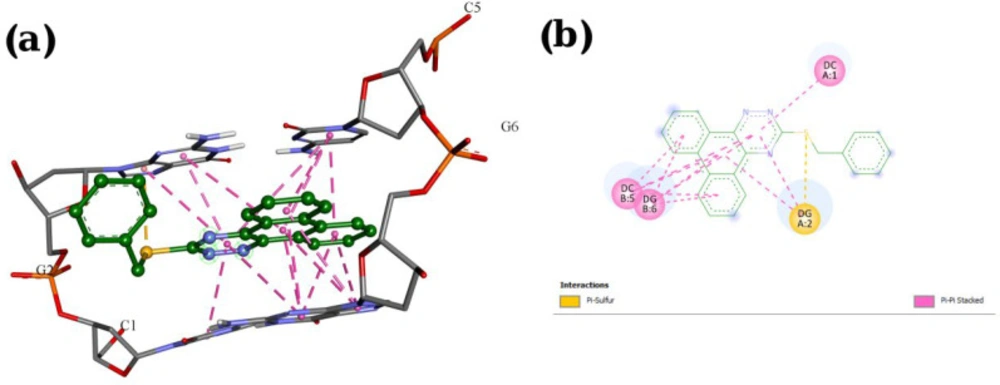
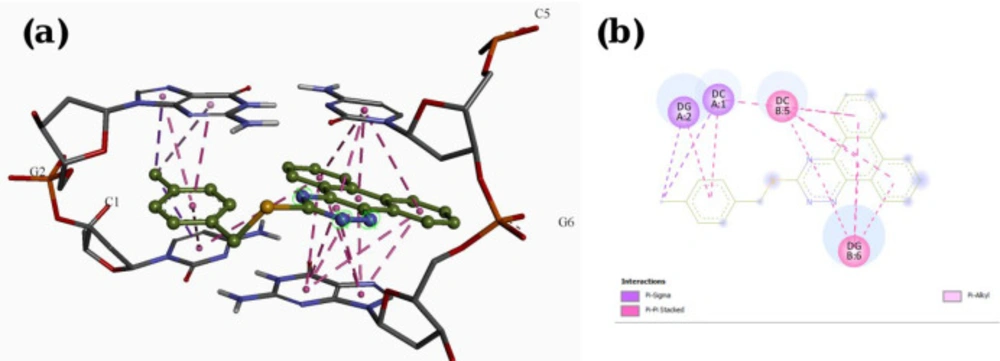
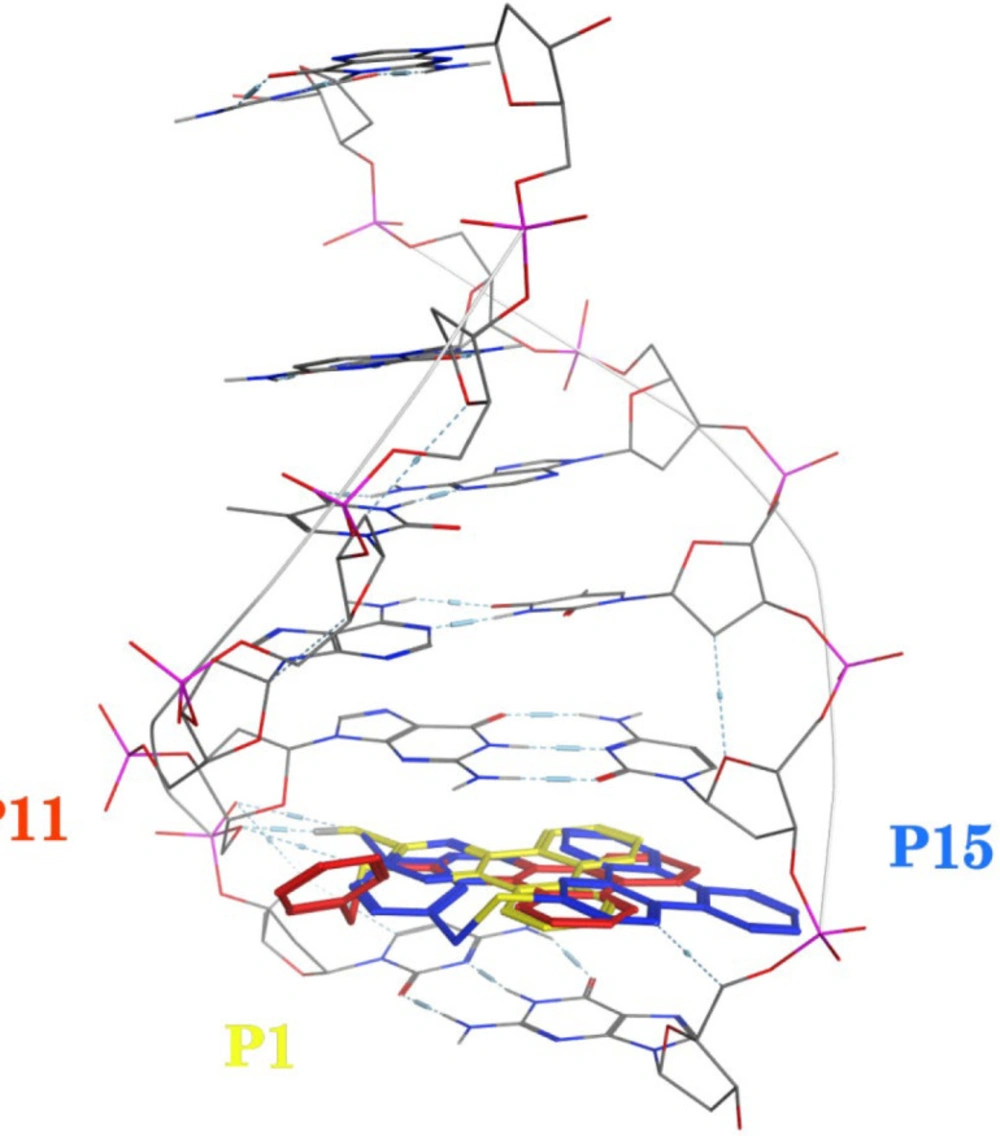
Molecular docking studies with DNA
The X-ray crystal structure of B-DNA (PDB ID: 1Z3F) was obtained from the Protein Data Bank. The ligand structures were drawn and minimized under Semi-empirical AM1 methods using HyperChem 7. Molecular docking studies were done using the AutoDock Tools 1.5.7rc1 and AutoDock 4.2.5.1 docking programs. The energy calculations were made using genetic algorithms (LGA) with the number of GA runs set to 100.
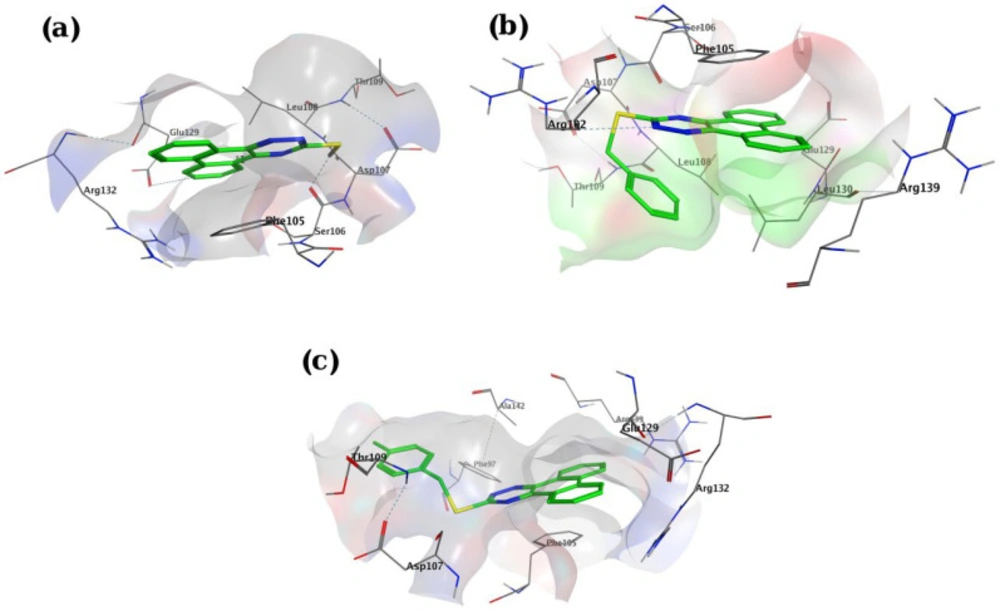
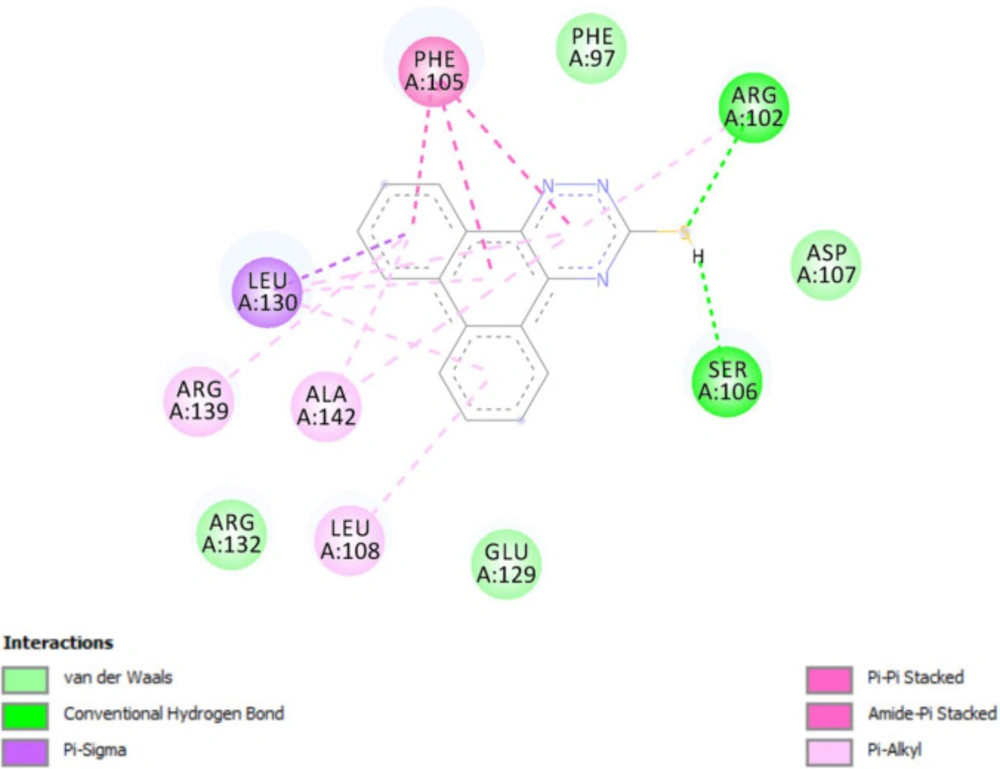
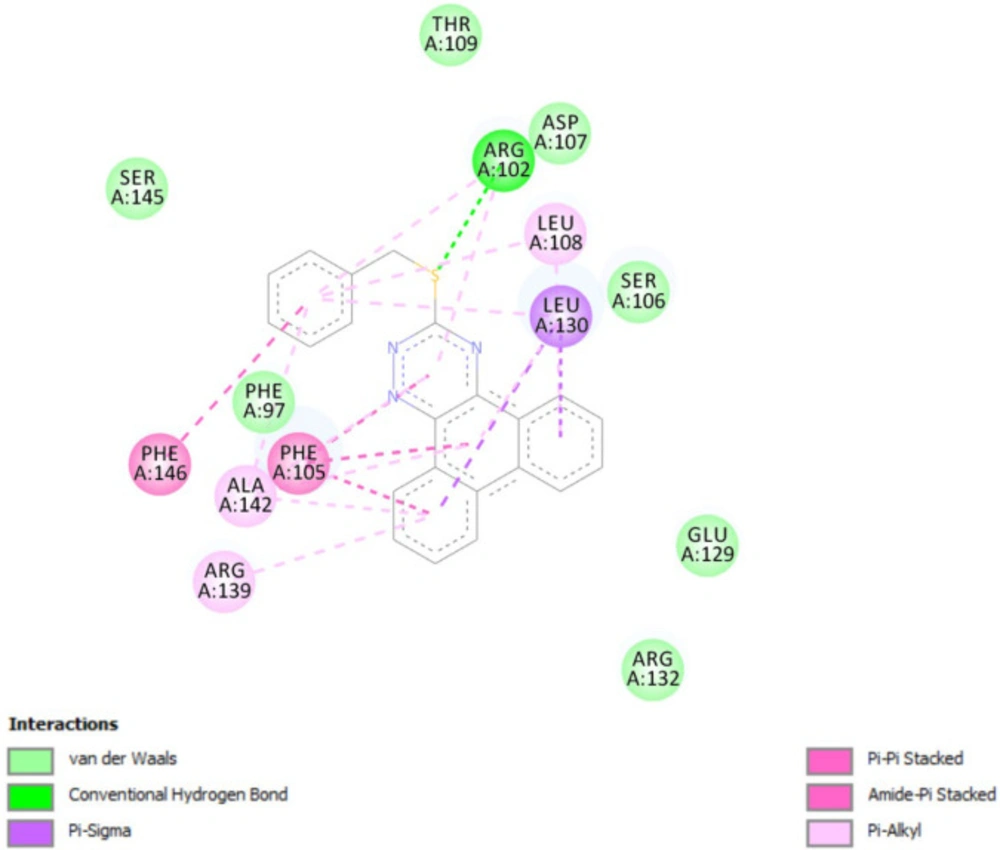
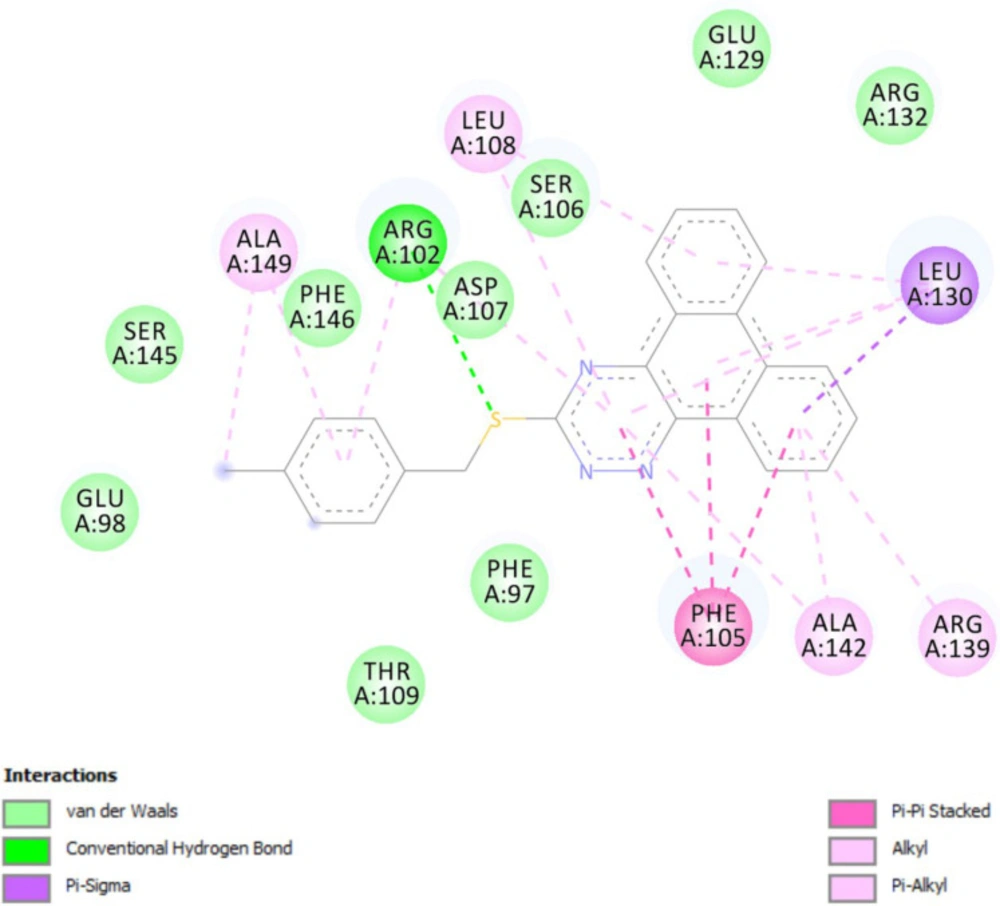
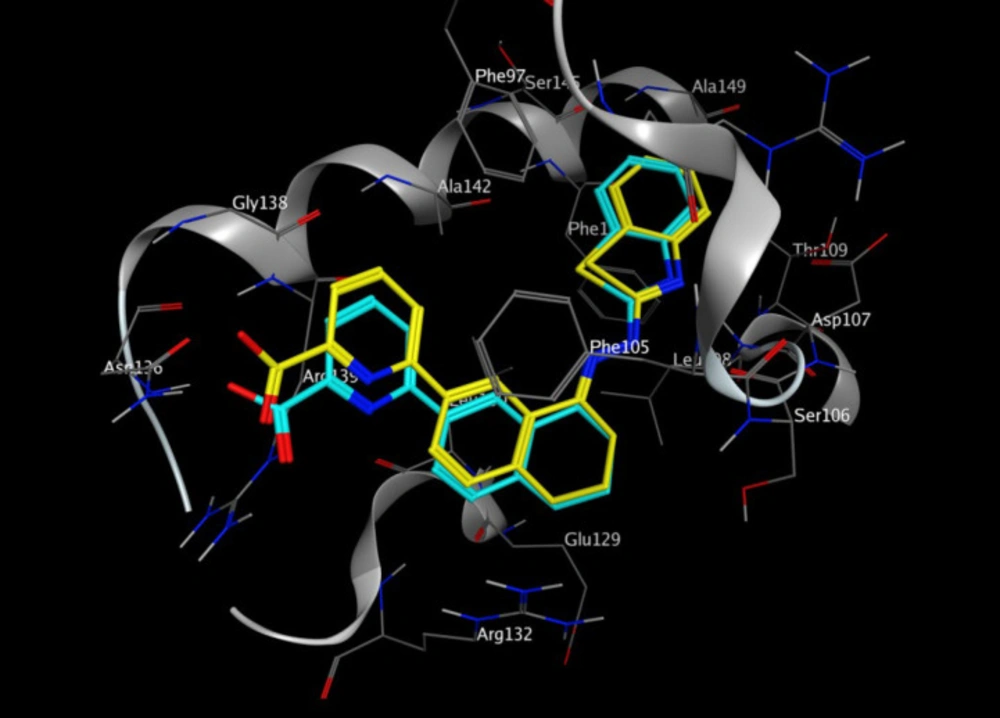
Molecular docking studies with Bcl-2 protein
The crystal structure of Bcl-2 (PDB ID: 3ZLN) was acquired from RCSB Protein Data Bank (http://www.rcsb.org).Innate ligand and waters were removed, hydrogen atoms were added, non-polar hydrogens were merged, and Gasteiger charges were added using AutoDock Tools to prepare the protein for docking. 3D structures of ligands were sketched and minimized under Molecular Mechanics MM+ and then Semi-empirical AM1 methods using HyperChem software. A grid of 60, 60, and 60 points in x = -17.788, y = -12.682 and z = 10.891 directions with 0.375 Å grid spacing was built. The docking process was performed by AutoDock 4.2. To find the conformers with the lowest binding energy, Lamarckian genetic algorithm (LGA) was used, and the number of GA runs was set to 100. The other parameters were left at program default values. Docking procedure validity was examined by re-docking the structure co-crystallized ligand into the receptor (self-docking) using the protocol mentioned above.