
Introduction
Human clotting factor IX (hFIX) is a Vitamin K-Dependent (VKD) plasma glycoprotein, which takes part in the middle stage of the intrinsic pathway of blood coagulation (1, 2). It is synthesized by hepatocytes and secreted as a serine protease zymogen into the bloodstream. The circulating hFIX zymogen consists of 415 amino acids which are divided into four identifiable structural domains. The Gla domain (residues 1-45), with high affinity to Ca2+ ions, is located at the N-terminus of the mature hFIX. In the presence of the Ca2+ ions, the Gla domain undergoes structural changes, acquires stable conformation, and mediates the interaction of hFIX with phospholipids. The Gla domain is followed by two epidermal growth factor (EGF)-like domains (EGF1 and EGF2). A trypsin-like serine protease domain is located at the C-terminus of the hFIX, which is connected to the EGF2 via a 35-residue activation peptide (3-6).
Proteolytic cleavages at Arg145-Ala146 and Arg180-Val181 peptide bonds by either factor XIa (FXIa) or factor VIIa/tissue factor (FVIIa-TF) complex, in the presence of Ca2+ ions result in releasing of the activation peptide and subsequent formation of the physiologically active hFIX (hFIXa) (7). The hFIXa interacts with its co-factor, activated factor VIII (FVIIIa), in a Ca2+-dependent manner to form a tenase complex on the phospholipid membrane surface to catalyze the conversion of factor X (FX) to its active form (FXa) (8).
Deficiency or malfunctioning of hFIX leads to an X-linked congenital bleeding disorder, hemophilia B, with an incidence rate of one in 30000 live male births (5). Replacement therapy is the major treatment for the hemophilia B which is carried out via infusion of either plasma derived or recombinant hFIX (9). However, in recent years, concerns over the possible transmission of human blood-born infectious agents have motivated the use of recombinant forms of biotherapeutic products (10, 11). The structural complexity of multiple post-translational modifications (PTMs) such as VKD γ-carboxylation, β-hydroxylation, O-linked and N-linked glycosylation necessitates that the recombinant form of hFIX to be expressed in mammalian cells (12, 13). Different mammalian expression systems have been used to produce recombinant hFIX to ensure its appropriate PTMs, similar to those that occur in human cells (13, 14). Currently, the CHO cell line is preferably used for the production of therapeutic glycoproteins, including the hFIX. This cell line offers several advantages, in particular, production of glycoproteins with N-glycan similar to those found in human glycoproteins (15).
N-glycosylation is a co-or post-translational process that occurs on most of the secretory and membrane proteins in eukaryotic cells in the endoplasmic reticulum (ER) (16, 17), where the glucan moiety attaches to the Asn residue side chain located within the N-X-S/T sequon (where X can be any amino acid except proline) (17-19). N-linked glycans are an important part of glycoproteins and have a profound effect on protein properties such as folding, transmission, stability, solubility, degradation, and eventually protein secretion (16, 18 and 20).
Glyco-engineering is a molecular approach to introduce new N-glycan(s) through the generation of new N-glycosylation site(s) within the amino acid sequences of the target proteins (21, 22). This process leads to hyper-glycosylation of the proteins and is mainly applied for the improvement of products of important biopharmaceuticals. Hyper-glycosylation may not only improve the pharmacokinetics of a therapeutic protein but in some cases, can increase the protein secretion by assisting its folding, especially when it occurs at C- or N-terminal region of a protein (18, 23 and 24). It has been demonstrated that secretion of a glycoprotein with an N-glycan close to the N-terminus can be improved by skipping the interaction with Bip and directly entering into the calnexin/calreticulin cycle (25). Secretion efficiency of a protein can be inversely correlated with the extent of Bip association in such a way that unglycosylated proteins display strong and stable associations with Bip (25, 26).
In this work, to investigate the impact of the hyper-glycosylation on the functional expression of the recombinant hFIX in CHO-s cells, the hFIX-encoding DNA was subjected for site-directed mutagenesis to introduce additional N-glycosylation sequon, somewhere in the N-terminal Gla domain, close to the protein N-terminus.
The candidate sites for hyper-glycosylation were selected based on the data obtained previously by molecular dynamic simulations of the 3D structure of the Ca2+ stabilized hFIX Gla domain (27). In this regard, two hFIX N-glycosylation mutants, namely; K22N and R37N were subjected for further expression analyses after being constructed via site-directed mutagenesis. Hyper-glycosylation occurrence, of the examined mutants and its impact on the functional expression of the hFIX was investigated at the various expression levels, including; transcription, translation, and post-translation.
Experimental
Materials and Methods
Site-directed mutagenesis and Construction of the recombinant expression plasmids
Based on overlap extension PCR (Figure 1), site-directed mutagenesis was applied to introduce mutations into the hFIX cDNA (28, 29). Oligonucleotides used for site-directed mutagenesis are listed in Table 1. Oligonucleotide pairs K22N-F/K22N-R and R37N-F/R37N-R were used respectively for introducing K22N and R37N substitutions into hFIX cDNA. Oligonucleotides hFIX-EcoRI and hFIX-BamHI equipped respectively with EcoRI and BamHI restriction sites, were used as forward and reverse primers for amplification of the pre-pro hFIX or its N-glycosylation mutants’ coding sequences. Primer hFIX-BamHI was equipped with a nucleotide sequence of 6xHis before the stop codon in order to introduce a His-tag to the recombinant proteins’ C-termini. The amplified fragments were then cloned into the pIRES2-EGFP vector, between EcoRI and BamHI restriction sites, upstream to the IRES-EGFP sequence.
Following NheI/NotI digestion, the target fragments, comprising the coding sequences of either the hFIX or its mutants and IRES-EGFP sequence, were subcloned into the pCEP4 expression vector (Thermo Fisher Scientific, USA), downstream to the cytomegalovirus (CMV) immediate early promoter/enhancer to end up with three bicistronic recombinant plasmids namely; pCEP4-hFIXwt, pCEP4-hFIXK22N, and pCEP4-hFIXR37N.
Cell Culture, Transfection, and Transient Expression
Suspension adapted Chinese hamster ovary (CHO-s) cell line (Thermo Fisher Scientific, USA), considered as a mammalian expression host, was cultured in FreeStyle™ CHO Expression Medium (Thermo Fisher Scientific, USA) supplemented with 8mM L-glutamine (Thermo Fisher Scientific, USA) and incubated at 37oC in an 8% CO2 atmosphere while shaking at 140 rpm. For transient expression, the cells were transfected with bicistronic recombinant plasmids; pCEP4-hFIXwt, pCEP4K22N, and pCEP4R37N (expressing either wild-type or mutant forms of the hFIX, together with an enhanced green fluorescent protein, EGFP, as a reporter protein), using FreeStyle™ MAX reagent (Thermo Fisher Scientific, USA), according to the manufacturer’s instruction. Three to five h after transfection, vitamin K (Caspian Tamin, Iran) was added at a final concentration of 2.5µg/mL. At 24, 48, and 72 h post-transfection, the cells were pelleted and the culture media were harvested and subjected for expression analyses.
Transcript Stability analysis
Secondary structures of the mRNAs from complete sequences of the hFIXwt and its N-glycosylation mutants’, were predicted using Mfold Web Server (http://unafold.rna.albany.edu/?q=mfold) (30). The analyzed regions included the coding sequences of the hFIXwt or its mutants and the IRES-EGFP sequence.
RNA Extraction and cDNA Synthesis
Total RNAs were isolated from the transfected CHO-s cells at 24, 48, and 72 h post-transfection using high pure RNA isolation Kit (Roche, Germany), according to the instruction provided by the manufacturer. Both quantity and purity of the total isolated RNAs were determined by UV absorbance measurement at 230, 260, and 280 nm by NanoDrop spectrophotometer. OD260/OD280 ratio of ~2 and OD260/OD230 ratio of 2-2.2 were considered as an acceptable purity for the RNA samples.
The integrity and quality of the purified RNAs were further checked using 2% agarose/formaldehyde electrophoresis and ethidium bromide staining. Then 1 µg of total RNA was treated with DNase I to eliminate genomic DNA and plasmids contaminations, and subjected for the first strand cDNA synthesis using oligo dT primers and revertAid M-MulV reverse transcriptase (Thermo scientific, USA).
Real-Time Quantitative PCR (qPCR)
The synthesized cDNAs were then used as a template for the subsequent qPCR experiment using hFIX-specific oligonucleotide hf9-RTF1 and hf9-RTR1 as forward and reverse primers, respectively (Table 1). A 117 bp fragment from hamster glyceraldehyde 3-phosphate dehydrogenase (Gapdh; a housekeeping gene) gene was amplified as an internal control using hams-gapdhF1 and hams-gapdhR1 primers (Table 1). To quantify the relative mRNA level of each construct, SYBR green-based real-time PCR was carried out using RealQ Plus Master Mix Green (Ampliqon, Denmark) on Corbett Rotor-Gene 6000 Real-Time PCR machine (Qiagen, USA). The recombinant gene transcription was normalized to that of the Gapdh gene. About 50 ng cDNA sample, 0.5 μL of each forward and reverse primer (10 µM) and 5 μL RealQ Plus Master Mix Green (Ampliqon), in a final volume of 10 μL, were used for real-time PCR. The qPCR amplification temperature profile was as follow: 95 °C for 5 min (denaturation), followed by 40 cycles of amplification for 20 s at 95 °C, 20 s at 61 °C, and 20 s at 72 °C. All qPCRs were performed in triplicates. A reaction sample without cDNA was performed as a control in each run. Gapdh housekeeping gene was used as an internal control for normalization. Specificity of the primer pairs and the purity of real-time PCR products were confirmed by melting curve analysis of 65 °C to 95 °C, with fluorescence measured every 0.5°C at the end of each reaction (with no nonspecific and primer-dimer formation). Gel electrophoresis was performed to approve the PCR products and primer specificity.
Flow Cytometry Analysis
CHO-s cells transfected with bicistronic recombinant plasmids, expressing hFIXwt or its N-glycosylation mutants along with EGFP as a reporter protein, were observed using fluorescence microscope 24, 48, and 72 h after transfection to detect EGFP signals. The transfection efficiencies were then monitored using flow cytometric analysis. About 2 × 105 CHO-s cells were harvested by centrifugation at 100 ×g for 5 min, and washed twice with PBS. The cytosolic expression of EGFP in transfected cells were analyzed using flow cytometry (BD FACSCalibur, USA). EGFP fluorescence was excited at 488 nm, and emission was measured with a 530/30 nm bandpass filter. Untransfected cells were served as a control for autofluorescence and were used to set the gate for positive cells to exclude debris. The results were analyzed using FlowJo software (Treestar, Inc.).
Quantification of the Recombinant hFIX Accumulated Inside the CHO-S Cells
After 72 h of transfection, 5 × 106 CHO-s cells were pelleted by centrifugation at 100 g for 5 min. The pellets were then re-suspended in 500 μL of ice-cold lysis buffer (50 mM Tris, pH 8, 150 mM NaCl, 0.1% triton X-100, 0.5% sodium deoxycholate, 0.1% SDS, containing protease inhibitor mix (Roche). After 20-30 min, the lysate was centrifuged at 12000 ×g for 20 min at 4 ºC, and the supernatant was analyzed for intra-cellular hFIX antigen, using ELISA.
Enzyme-Linked Immunosorbent Assay (ELISA) of hFIX Antigen (hFIX::Ag) in Culture Media
Culture media were subjected for quantification of hFIXwt or its N-glycosylation mutants using standard ELISA, based on the procedure provided by the manufacturer (Asserachrom, France). To quantify intracellular expression of recombinant hFIXwt or mutants, 50 µL of cell lysate was used for ELISA assay. The concentration of the expressed hFIXwt and mutants in culture media or intra-cell were calculated based on the standard curve and stated in ng/106 transfected cells.
Coagulation Assay
The coagulation activity of the secreted hFIXwt and its N-glycosylation mutants were measured using a chromogenic assay, according to manufacturer’s protocol (Biophen factor IX kit, France). The citrated human plasma derived from normal individuals was used as a standard sample. The culture medium from untransfected cells was used as a negative control.
The hFIX Purification
Recombinant proteins expressed by CHO-s cells transfected with bicistronic expression plasmids were then purified using Ni-NTA purification system. After 72 h of transfection, the cell suspensions were centrifuged at 100 g for 5 min and culture media were harvested. About 15 mL of the culture media was subjected for affinity purification using Ni-NTA beads (provided by Dr. Mohammadi at NIGEB, Iran).
SDS-PAGE and Western Blotting
The purified recombinant hFIX and mutants were analyzed using 4–12% gradient SDS-PAGE (31) to characterize hyper-glycosylated mutants in comparison with the native hFIX. Electroblotting of proteins onto polyvinylidene difluoride (PVDF) membrane (Roche, Germany) was performed using a wet procedure, in a transfer buffer (25 mM Tris, 192 mM glycine, 20% methanol) for 3 h at 300 mA.
The blot was then blocked in 5% non-fat milk solution (5% w/v, in Tris-buffered Saline containing 0.1% Tween 20) and subjected for Western blotting experiment during which the electroblotted protein bands were probed with a rabbit polyclonal anti-hFIX primary antibody (Abcan, UK) with dilution of 1/2500 which was followed by incubation with goat anti-rabbit secondary antibody conjugated with peroxidase (Millipore, Co. US) with a dilution of 1/2500. Immunoreactive bands were visualized by enhanced chemiluminescence (ECL) Western blotting detection kit (Najm, Iran) or by 4-chloronaphtol substrate.
Enzymatic Deglycosylation
The purified wild-type and mutant rhFIX samples were subjected for deglycosylation using PNGase-F enzyme kit (Biolabs, New England) according to the manufacturer’s protocol. Following the denaturation step, the protein samples were treated with PNGase-F enzyme in reaction buffer containing 1% NP-40. Deglycosylated samples were then examined by 4-12% gradient SDS-PAGE and Western blotting analysis.
Statistical Analysis
All experiments were done in triplicates. The values were expressed as means±SD. The statistical analyses were performed by one-way analysis of variance (ANOVA) followed by Duncan post hoc using SPSS version 22 software. The P values less than 0.05 were considered to be statistically significant.
Results
Transfection Efficiency Analysis
The three bicistronic plasmids, expressing either the wild-type or one of the two N-glycosylation mutants (hFIXK22N and hFIXR37N(, along with EGEF, were separately applied for transient transfection of CHO-s cells. The transfection efficiency was evaluated using flow cytometry at 24, 48, and 72 h post-transfection. The untransfected cells were used as a control to exclude autofluorescence (Figure 2). All the examined hFIX mutants contained the hFIX native signal peptide; therefore, they were expected to be secreted into the culture media, whereas the expressed EGFP remained in the cytosol, and the cells appeared in green color under fluorescence microscopy (Figure 2).
The hFIX Transcript Analysis
Transcription of the recombinant hFIXwt and its mutants in transiently transfected CHO-s cells were confirmed by performing RT-PCR and qPCR, at different post-transfection time points (24, 48, and 72 h). Electrophoresis pattern of the amplified cDNA by hFIX specific primers yielded a PCR product of 117 bp. Hamster’s Gapdh gene was also amplified using internal specific primers to produce 116 bp PCR products.
In the continuation of the transcripts’ analysis, comparative qPCRs were performed on the mutants to investigate the variation of hFIX mRNA levels due to the mutations (Figure 3). Normalization was carried out using amplification of hamster’s Gapdh gene. As the results revealed, the transcription level for hFIXwt was significantly higher than those of the examined mutants at 24 h post-transfection. In fact, the highest transcription expression level occurred for the wild-type construct 24 h after transfection. No significant differences between the mutants at transcription expression levels were observed (Figure 3).
Immunoassay of the Expressed hFIX Mutants
Considering that N-glycosylation often drastically increases the molecular weight of a protein, the two hFIX N-glycosylation mutants and the hFIXwt were subjected for gradient SDS-PAGE and Western blotting analysis after purification using Ni-NTA beads. The three samples were detectable with the specific anti-hFIX antibody. The hFIXR37N showed a higher molecular weight than the hFIXwt and hFIXK22N (Figure 4), suggesting the probability of the hyper-glycosylation occurrence in the case of the hFIXR37N.
In order to confirm the correlation between the increased molecular weight of the hFIXR37N and N-glycan attachment, the hFIXR37N along with the hFIXwt were treated with PNGase-F and examined using gradient SDS-PAGE and Western blotting analysis (Figure 5). PNGase-F is an amidase that catalyzes the cleavage between the innermost GlcNAc and asparagine residues from N-linked glycoproteins (32). As shown in the Western blotting results, both samples migrated at the same molecular weight after being treated with PNGase-F (Figure 5). Indeed, an expected gel-shift upon similar treatment of both wild-type hFIX and hFIXR37N mutant with PNGase-F was observed and the same protein size on the blot was obtained (Figure 5, lanes 2 and 4).
Assessment of the hFIX Expression Level Based on the ELISA In order to examine the protein expression levels of the wild-type hFIX and its mutants, during 72 h after transfection, the culture media were harvested at three time points, 24, 48, and 72 h post-transfection, and subjected for hFIX expression analysis using ELISA. Based on the results obtained, the wild-type hFIX and mutants were all recognized by the hFIX antibody. At 48 and 72 h post-transfection, both mutants showed a significant increase in the hFIX expression level, when compared with that of the hFIXwt (Figure 6). However, the highest hFIX expression levels for all three constructs occurred on the 3rd day after transfection.
Evaluation of Secretion Efficiency of the Expressed hFIXwt and mutants
Secretion efficiency of a protein is the ratio between the secreted fraction of the protein and its total amount (secreted plus intracellular fractions) (33). In order to calculate the hFIX secretion efficiencies for the examined constructs, their corresponding cell lysates and culture media were harvested 72 h after transfection and considered for secretion efficiency assessment. Analysis of the hFIX level in cell lysates and calculation of secretion efficiencies of the examined constructs indicated no significant differences between secretion efficiencies of the mutants and their wide-type counterpart (Figure 7).
Measurement of Coagulation Activity of the Secreted hFIX
The coagulation activities of the hFIXwt and N-glycosylation mutants in the culture media were measured by chromogenic assay at different time points. Our results showed a reduced clotting activity for the hFIXR37N mutant, compared to that of the hFIXwt (Figure 8). No significant difference was observed between clotting activities of the hFIXK22N mutant and the hFIXwt (Figure 8).
The Prediction of the Transcript Stabilities
The mRNA stabilities for the wild-type and mutants in the form of bicistronic mRNAs were obtained through calculating free ΔG. The free ΔGs for three mRNA were -913.20, -915.70, and -915.40 for hFIXwt, hFIXK22N, and hFIXR37N respectively, which showed no significant differences among them.
Discussion
Based on the previously described rational methodologies (27), two positions were selected to introduce new N-glycosylation sites in the Gla domain of the hFIX by means of site-directed mutagenesis and single amino acid substitution. Introducing a new N-glycosylation site does not ensure the glycan attachment to Asn residue located in there (18). In order to investigate the N-glycosylation occurrence in the newly introduced N-glycosylation sites of two hFIX mutants, the culture media from the transfected cells were collected and subjected for gradient SDS-PAGE. The hFIXR37N mutant showed higher molecular weight that the hFIXwt, implying that hyper-glycosylation might occur, while the hFIXK22N mutant demonstrated the same molecular weight as the wild-type. After PNGase treatment of the hFIXR37N in parallel with the wild-type, the hFIXR37N mutant acquired the same size as that of the hFIXwt. This finding confirmed the creation of a new N-glycosylation site in the hFIXR37N mutant, recognizable by oligosaccharyl transferase for an N-glycan attachment to Asn37 residue.
Failure of hyper-glycosylation in the case of the hFIXK22N mutant, can be explained by the participation of the Cys23, located at +1 position in the newly generated N-glycosylation site (i.e. Asn22-Cys23-Ser24), in a disulfide bond formation with the Cys18, generating a loop (34, 35). The generated loop, which entraps the N-glycan acceptor Asn22, might prevent the attachment of the oligosaccharide to Asn22 residue. Previously it has been reported that N-glycosylation and disulfide bond formation have reciprocal effects so that a glycan attachment may sterically hinder the formation of a disulfide bond, and a disulfide bond formation, in turn, may prevent the oligosaccharide attachment to the nearby N-glycosylation site (36). In an experiment carried out by McGinnes and coworkers on hemagglutinin-neuraminidase (HN), a glycoprotein of the Newcastle disease virus (paramyxovirus), either of the cysteine residues flanking an unused N-glycosylation site was removed by substitutional mutation, leading to the attachment of a glycan (37). In this experiment, site-directed mutagenesis was carried out to remove either of the two cysteine residues participating in the formation of a disulfide bridge near the unused N-glycosylation site. Their results indicated the attachment of oligosaccharide after the removal of the disulfide bond. They concluded that the disulfide bond formation nearby the N-glycosylation site might prevent the glycan attachment (37).
In the case of hFIXK22N mutant, a serine residue at position +2 in the Asn22-Cys23-Ser24 N-glycosylation-site, which is reported to be less effective than a threonine in the attachment of glycan to the Asn residue, might prevent the N-glycosylation (22).
Comparison of the expression efficiencies of the two hFIX mutants with that of the wild-type, at transcriptional and translational levels, led to different outcomes. At transcription level, the highest expression was related to the wild-type hFIXat 24 h after transfection, while at protein levels both mutants had higher expression than the wild-type at 48 and 72 h post-transfection. Since there were no significant differences among the ΔGs of the transcripts, the higher protein expression observed for both mutants cannot be attributed to the stability of their transcripts.
Our previous in-silico studies indicated in the increased stabilities for both mutants, hFIXK22N and hFIXR37N (27). In agreement with the in-silico studies, the higher protein expression levels were demonstrated for the two hFIX mutants in comparison with the hFIXwt, which might be due to their higher proteolytic stabilities, and their accumulations during the time courses of the protein production. In fact, the higher stabilities of the hFIX mutants in this work can be due to substitutions of Asn for Lys or Arg, in the hFIXK22N and hFIXR37N, respectively, and elimination of cleavage sites for a number of proteases including; LysC, LysN, trypsin, and Arg-C (38).
Comparison of the coagulation activity of the hFIXK22N mutant with that of the hFIXwt showed no significant difference. However, the hyper-glycosylated mutant, hFIXR37N, showed a reduced clotting activity when compared with that of the hFIXwt. This result indicated that the attachment of new N-glycan interferes with the enzymatic activity of the hFIX. The new N-glycosylation site in this hyper-glycosylated mutant is located within the protein Gla domain, which participates in the interaction of the protein with coagulation factor XI, another important protein in coagulation cascade, as well as phospholipid bilayer (39). It was shown that hyper-glycosylation of proteins in some cases increase the protein secretion by assisting its folding (18, 23 and 24). However, this was not the case for the hFIX mutants examined in this work. The effect of N-glycans close to the N- or C-terminal ends of a protein on its secretion efficiency is protein dependent and varies among different protein (18). Although relatively higher expression levels were observed for both of the hFIX mutants, regardless of their opposite outcomes in receiving an extra N-glycan, their secretion efficiencies were not affected by the mutation in CHO-s cells.
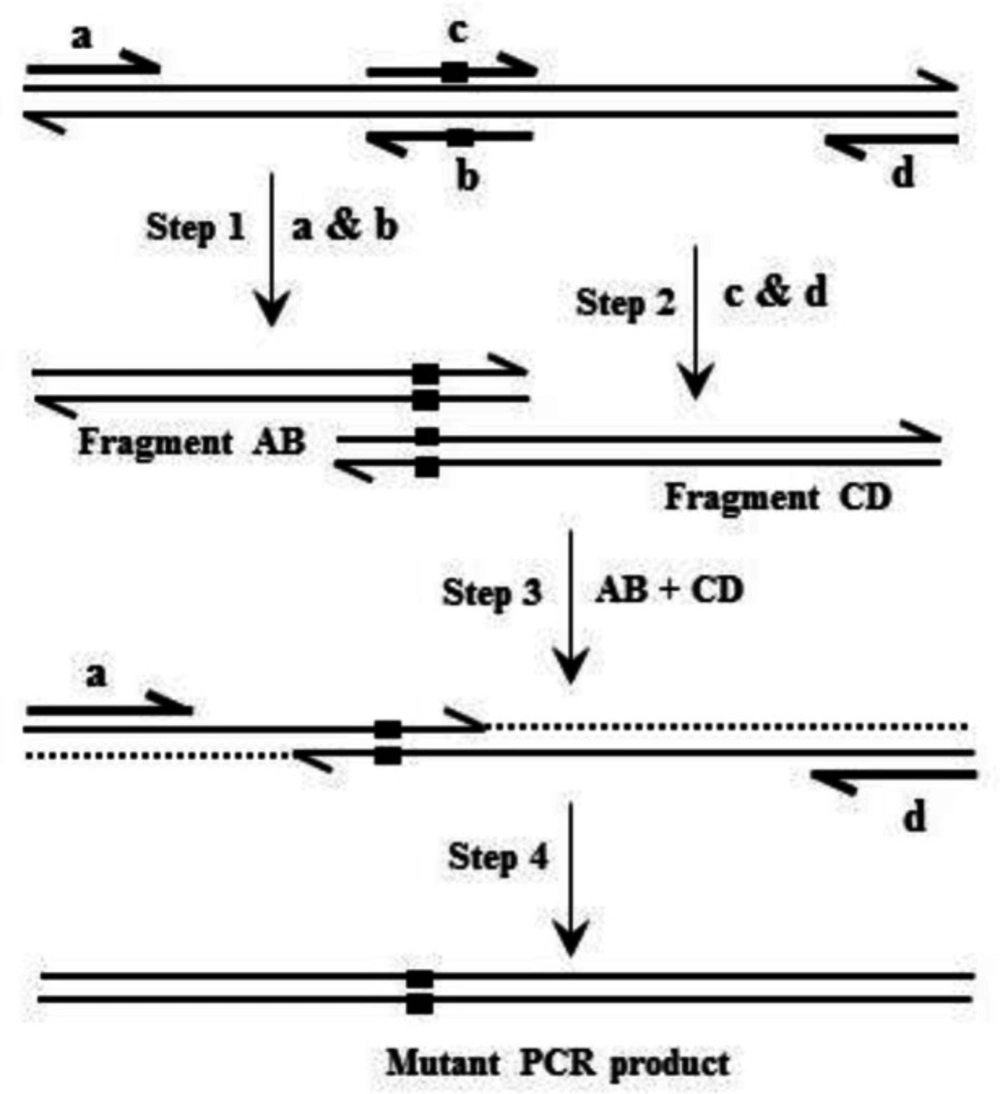
Schematic demonstration of the site-directed mutagenesis and construction of the hFIX N-glycosylation mutant expression plasmids
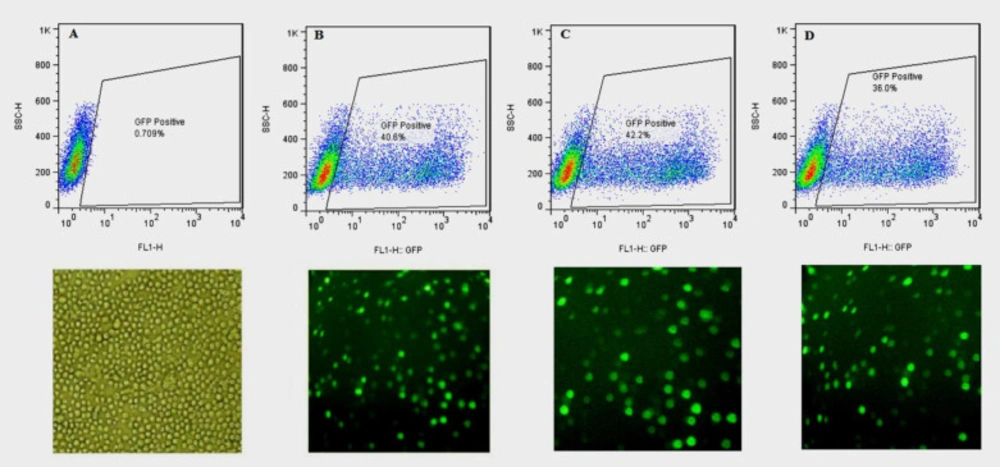
Photograph of the transfected CHO-s cells obtained with a fluorescence microscope,at magnification of 10X, and the rate of transfection obtained with flow cytometry,24 h after transfection. (A) Untransfected CHO-s cells, (B) CHO-s cells transfected with pCEP4-hFIXwt, (C) CHO-s cells transfected with pCEP4-hFIXK22N, (D) CHO-s cells transfected with pCEP4-hFIXR37N plasmids
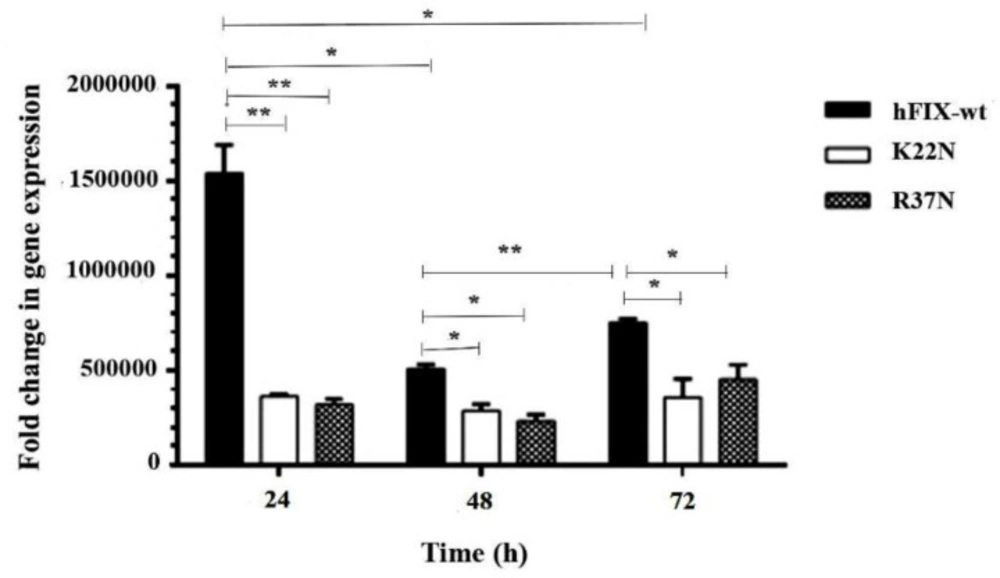
qPCR analysis of transcripts from hFIXwt and two N-glycosylation mutants 24, 48 and 72 h after transfection. Asterisks indicate samples that are significantly different (p < 0.05) compared to other samples, using analysis of variance. *p ≤ 0.05, **p ≤ 0.01

Western blotting analysis of the purified wild-type and mutant forms of hFIX expressed by transiently transfected CHO-s cells. Lane 1, protein molecular weight, Lane 2: hFIXwt, Lane 3, hFIXK22N mutant, Lane 5, and hFIXR37N mutant
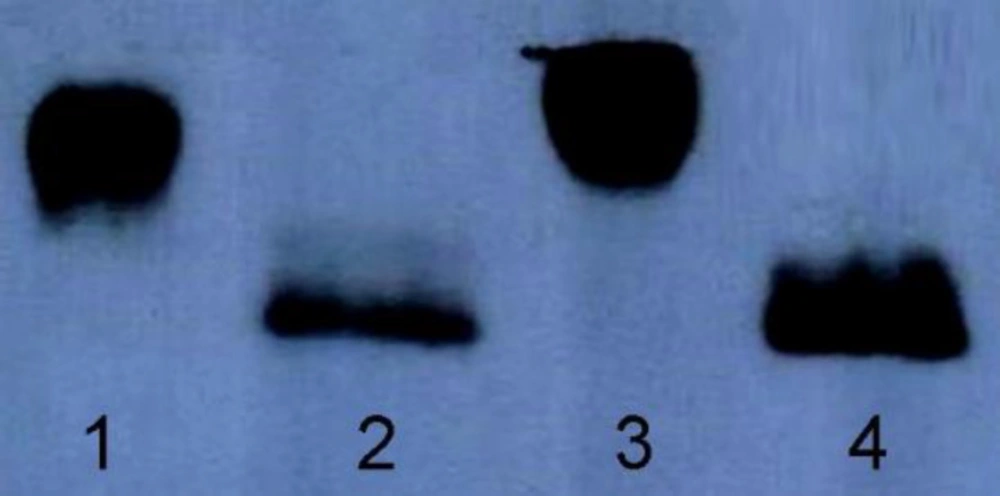
Western blotting analysis of the hFIXwt and the hFIXR37N before and after PNGase digestion. Lane 1: hFIXwt before digestion with PNGase, lane 2: hFIXwt after digestion with PNGase, lane 3: hFIXR37N mutant before digestion with PNGas, lane4: hFIXR37N mutant after digestion with PNGase
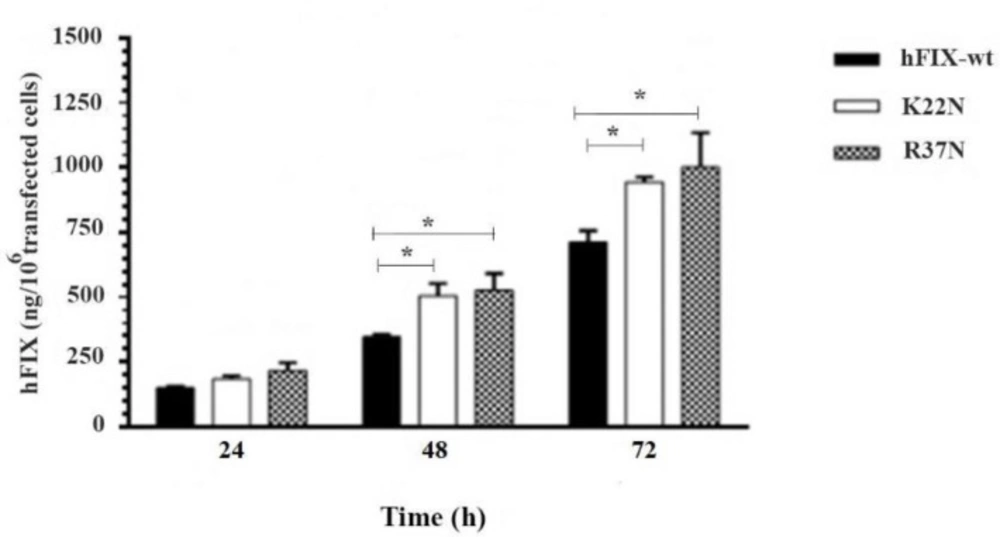
Evaluation of transient expression of hFIXwt and its N-glycosylation mutants, hFIXK22N and hFIXR37N, in CHO-s cells at various post-transfection time (h), based on ELISA performed on the samples taken from culture media. Asterisks indicate samples that are significantly different (p < 0.05) compared to other samples, using analysis of variance
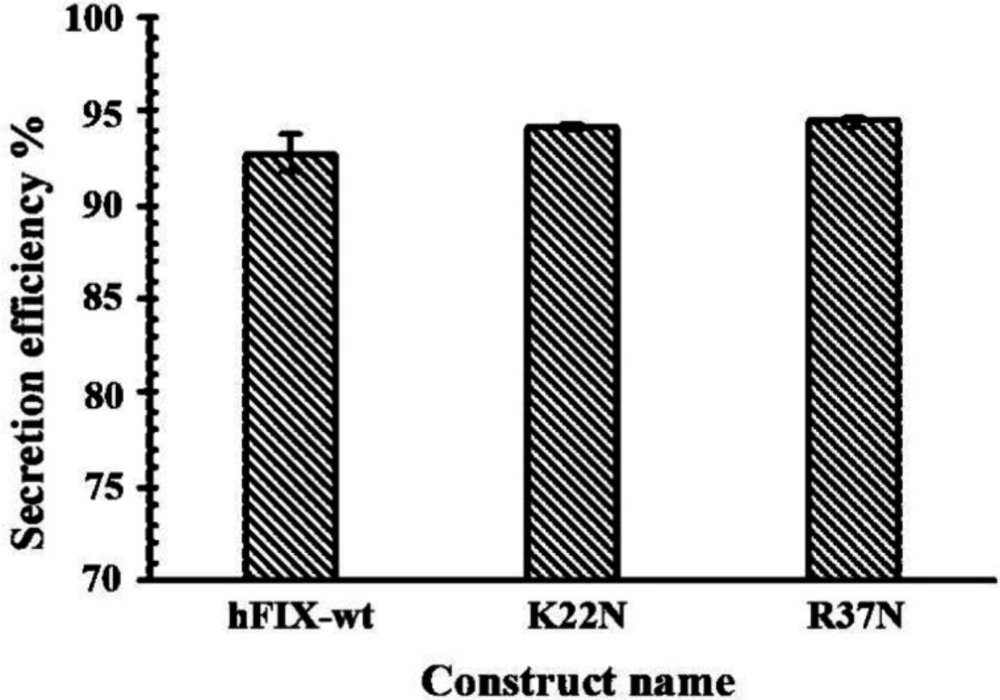
The hFIX Secretion efficiency by CHO-s cells 72 h post-transfection by hFIXwt, hFIXK22N and hFIXR37N constructs
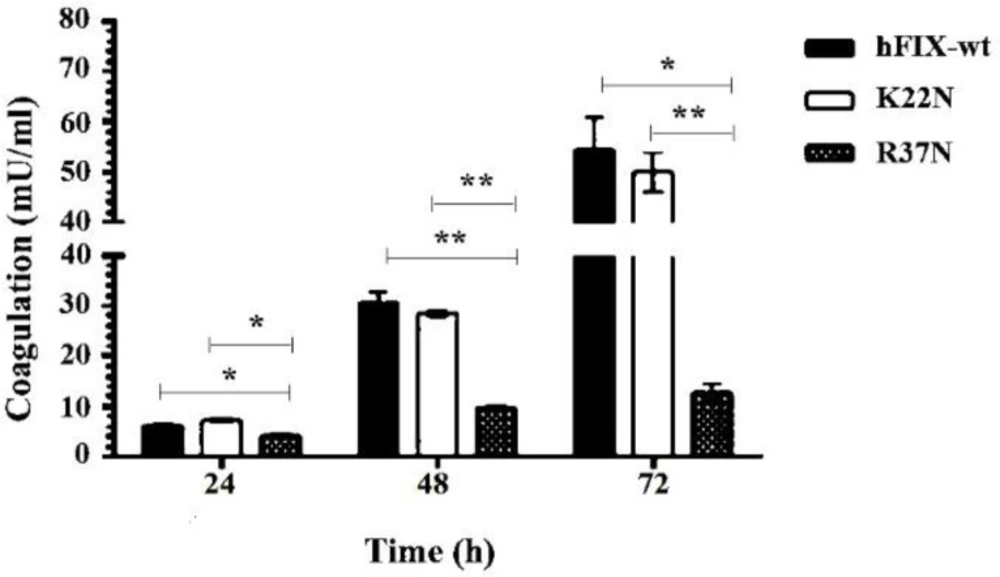
Coagulation assay for hFIXwt in comparison with the N-glycosylation mutants; hFIXK22N and hFIXR37N, during 72 h after transfection. Asterisks indicate samples that are significantly different (p < 0.05) compared to other samples, using analysis of variance. *p ≤ 0.05, **p ≤ 0.01
| primers | Nucleotide sequences (5´ → 3´) | Restriction site |
|---|---|---|
| hFIX-EcoRI | CCGGAATTC GCC ACC ATG CAG CGC GTG | EcoRI |
| hFIX-BamHI | CGCGGATCCTCATTAAGTGAGCTTTGTTTT | BamHI |
| K22N-F | TGTATGGAAGAAAACTGTAGTTTTGAAGAAGC | - |
| K22N-R | GCTTCTTCAAAACTACAGTTTTCTTCCATAC | - |
| R37N-F | GAAAACACTGAAAACACAACTGAATTTTGG | - |
| R37N-R | CCAAAATTCAGTTGTGTTTTCAGTGTTTTC | - |
| hf9-RTF1 | ||
| hf9-RTR1 | ||
| hams-gapdhF1 | ||
| hams-gapdhR1 |
The primes used for qPCR, mutagenesis and amplification of the coding sequences of the hFIXwt and its N-glycosylation mutants, the restriction sites are underlined
Conclusion
In accordance with our previous in-silico prediction, hyper-glycosylation of hFIX in the new N-glycosylation site, introduced by R37N amino acid substitution, was approved. Regardless of the N-glycan attachment in the case of both mutants, their expression in protein level was increased in comparison with the hFIXwt. No significant difference was observed between the free ΔGs of the transcripts, and the secretion efficiencies of the hFIX mutants in CHO-s cells were not affected by the mutations. Therefore, the improvement of the expression efficiencies of the hFIX mutants, at protein level could be attributed to enhanced protein stability, via omitting certain protease cleavage site. The hFIXR37N mutant showed a decreased clotting activity when compared with that of the hFIXwt, probably due to interference of the new N-glycan with the enzymatic activity of the hFIXR37N.
