
Introduction
Experimental
Results
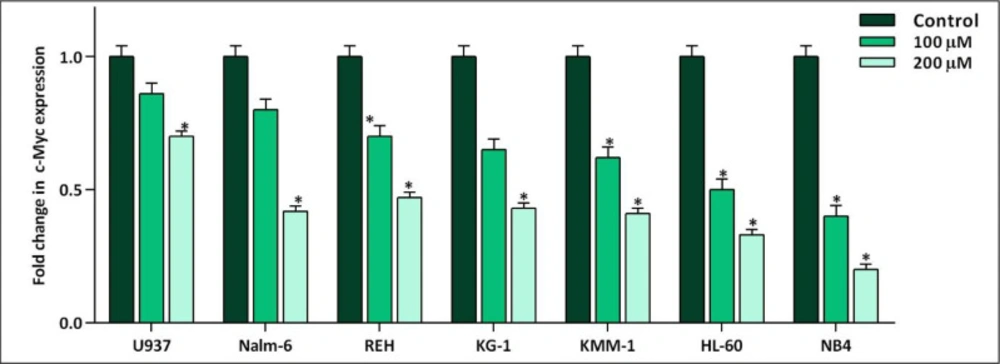
The effect of 10058-F4 on the expression level of c-Myc. After exposing the panel of hematologic malignant cell lines with 10058-F4 at the concentrations of 100 and 200 µM, the mRNA expression level of c-Myc was evaluated. Values are given as mean ± standard deviation of three independent experiments. *P ≤ 0.05 represented significant changes from the control
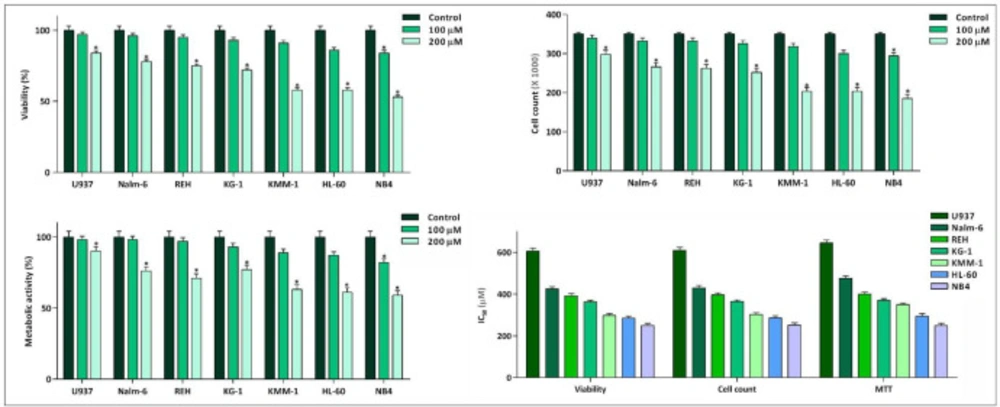
Inhibitory effect of 10058-F4 on viability, cell count, and metabolic activity of leukemic cell lines. 10058-F4 induced anti-leukemic effect in all cell lines; however, a different cell sensitivity pattern was noted among the tested cells. Values are given as mean ± standard deviation of three independent experiments. *P ≤ 0.05 represented significant changes from the control
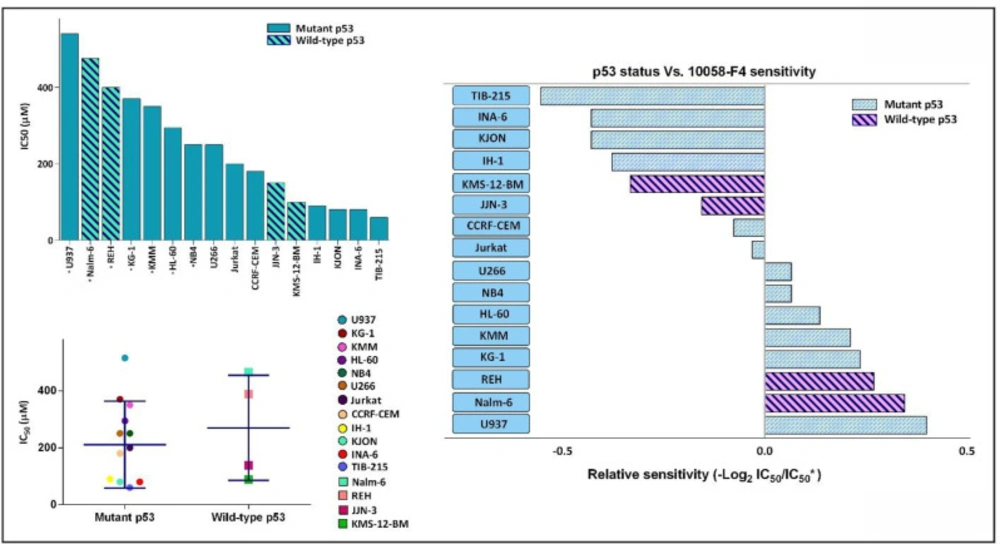
The anti-leukemic effect of 10058-F4 on hematologic malignant cell lines is exerted irrespective of the molecular status of p53. Based on our supplemental investigation and an extensive literature review, a list of IC50 response of different leukemic cell lines to 10058-F4 after 24 h was made. IC50 of starred cell lines was evaluated in our laboratory. Dot blot showing correlation between p53 status and in-vitro drug sensitivity as shown by the IC50 of individual cell line. Lines indicate median value. We failed to identify an obvious association between p53 status and leukemic cell sensitivity to 10058-F4. Values are given as mean ± standard deviation of three independent experiments
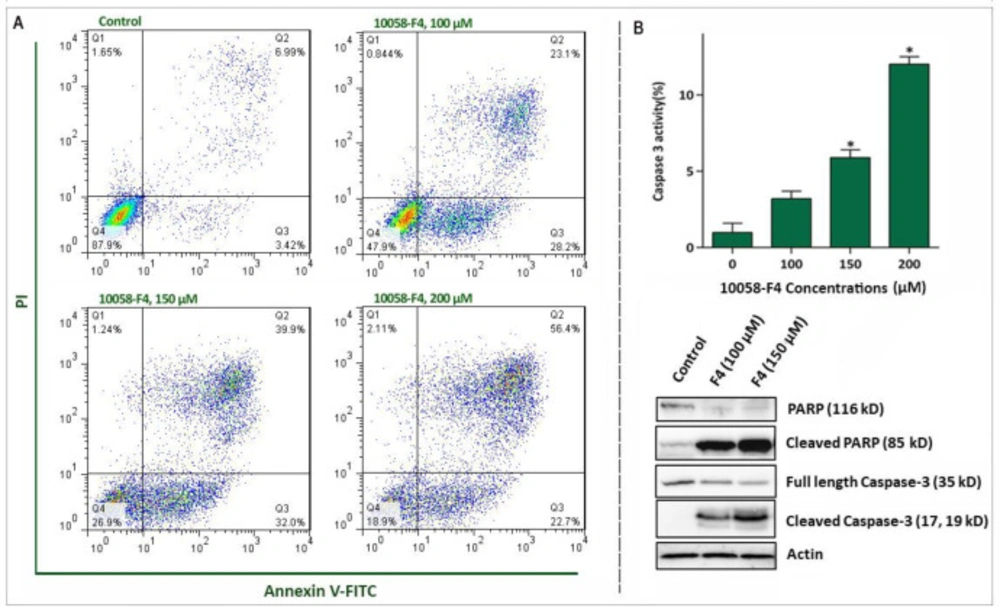
Abrogation of c-Myc induced caspase‐3‐dependent apoptosis in NB4 cell lines. (A) Treatment of NB4 cells with 10058-F4 remarkably increased the percentages of Annexin-V/PI positive cells. (B) 10058-F4 imposed a considerable elevation in caspase-3 activity and increased the amount of cleaved caspase 3 and PAPR. Values are given as mean ± SD of three independent experiments. *P ≤ 0.05 represented significant changes from the control
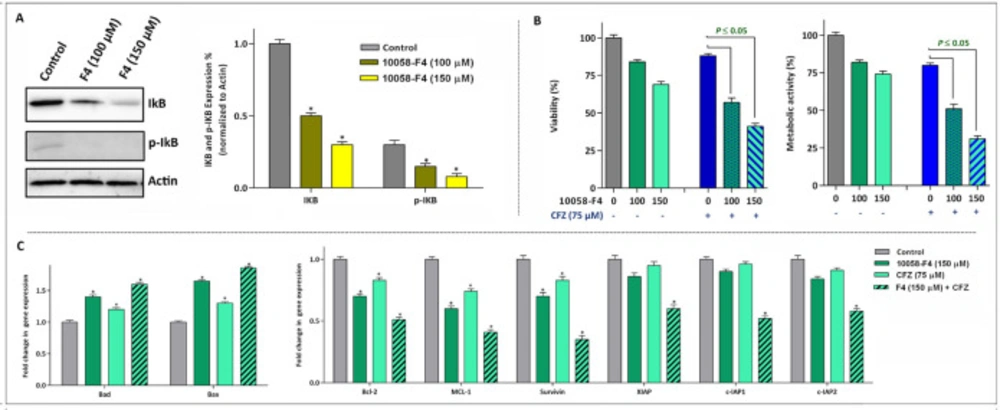
The anti-leukemic effect of 10058-F4 is mediated through suppression of NF-κB and its associated genes. (A) After the treatment of cells with the indicated concentrations of 10058-F4 for 24 h, total cell lysates were prepared and western blotting was performed using antibodies specific to IκB, p-IκB and Actin. (B) Suppression of proteasome using carfilzomib (CFZ) potentiate the anti-leukemic effect of 10058-F4. (C) The results of qRT-PCR analysis revealed that expression of both pro- and anti-apoptotic target genes altered more significantly when 10058-F4 was used in combination with CFZ. Values are given as mean ± SD of three independent experiments. *P ≤ 0.05 represented significant changes from the control
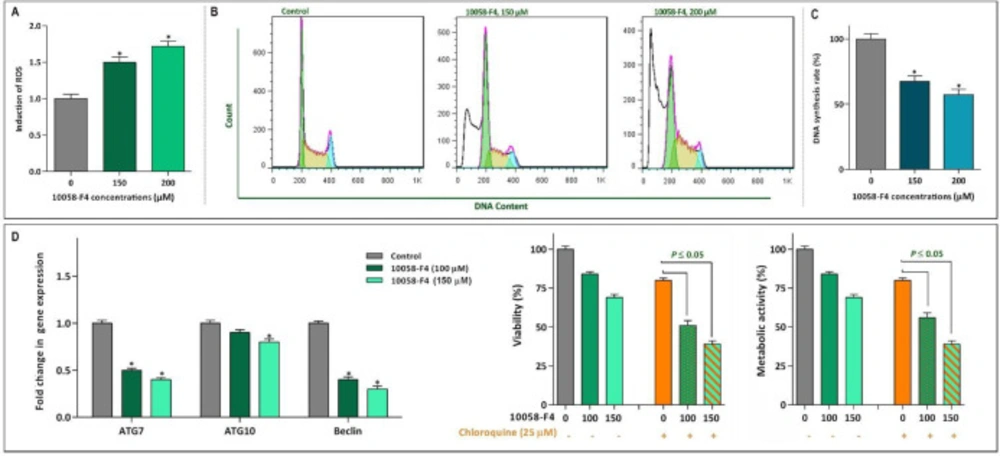
Effect of 10058-F4 on ROS level, the cell cycle progression and autophagy system. (A) Upon treatment of NB4 cells with 1008-F4, the intercellular level of ROS was elevated in a concentrations-dependent manner. (B) Treatment of the cells with 10058-F4 increased the proportion of NB4 cells in sub-G1 phase, while there was a reduction in the percentage of cells S phase. (C) The results of BrdU assay showed that 10058‐F4 could hamper the replicative potential of APL cells through reducing DNA synthesis rate. (D) The single agent of 10058-F4 could remarkably diminished the expression level of autophagy-related genes in NB4 cells. Moreover, when autophagy system was suppressed using chloroquine, the cytotoxic effect of 10058-F4 was remarkably reinforced. Values are given as mean ± SD of three independent experiments. *P ≤ 0.05 represented significant changes from the control
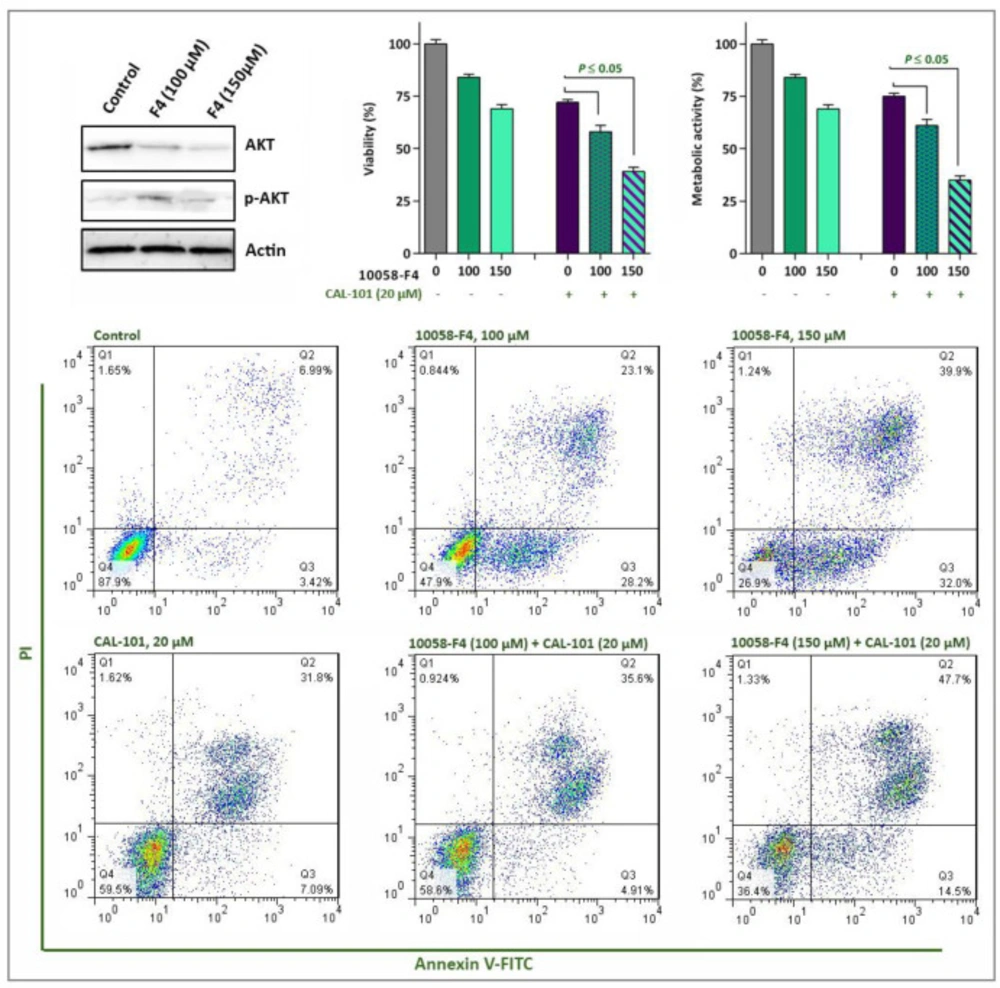
The effect of 10058-F4 on the PI3K signaling pathway. The results of western blot analyzing revealed that upon c-Myc inhibition the amount of phosphorylated Akt increased in NB4 cells. When 10058-F4 was accompanied by a PI3K inhibitor, CAL-101, the survival of NB4 cells were decreased more efficiently as compared to either agents alone. Values are given as mean ± SD of three independent experiments. *P ≤ 0.05 represented significant changes from the control
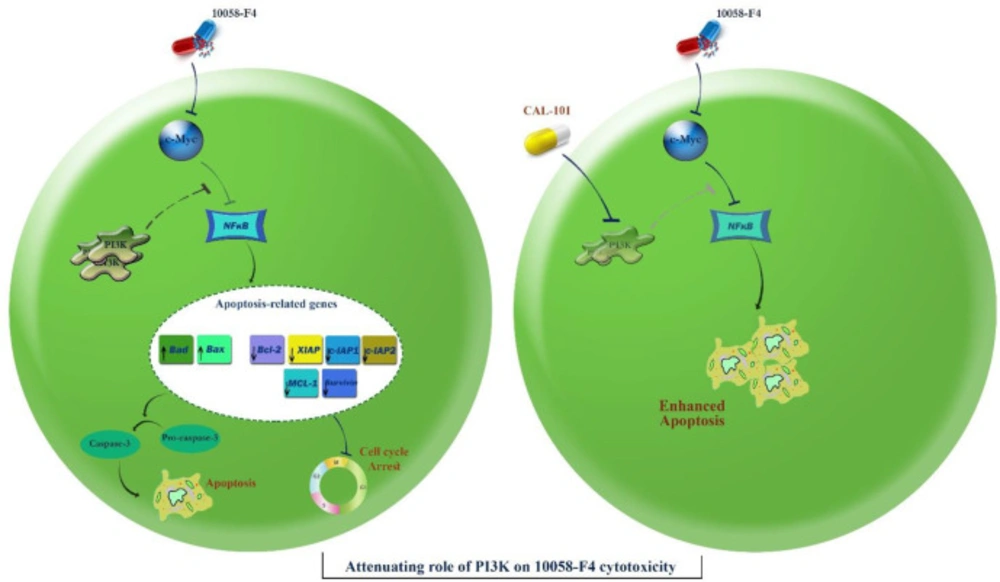
Schematic representation proposed for the plausible mechanisms of action of 10058-F4 in APL-derived NB4 cells. Through inhibition of c-Myc, 10058-F4 augmented the intracellular level of ROS and induced caspase-3-dependent apoptotic cell death in NB4 cells through suppression of the NF-κB signaling pathway. This favorable anti-leukemic activity could be attenuated through activation of the PI3K signaling pathway. Suppression of the PI3K axis using CAL-101 eliminated the compensatory effect of this pathway on 10058-F4-induced cytotoxic effect and promoted a more significant apoptotic cell death in NB4 cells
