Chemistry
All the chemicals, along with analytical grade solvents, were purchased from Sigma-Aldrich, Alfa Aesar (Germany), or Merck through local suppliers. Pre-coated silica gel Al-plates were used for TLC with ethyl acetate and n-hexane as solvent system (25:75). The spots were detected by UV254. Gallonkamp apparatus was used to detect melting points (uncorrected) in capillary tubes. IR spectra (ν, cm–1) were recorded by KBr pellet method in the Jasco-320-A spectrophotometer. EI-MS spectra were measured on a JEOL JMS-600H instrument with data processing system. 1H-NMR spectra (δ, ppm) were recorded at 600 MHz (13C-NMR spectra, at 150 MHz) in DMSO-d6 using the Bruker Advance III 600 As- cend spectrometer using BBO probe. The coupling constant (J) is given in Hz and chemical shift () in ppm. The abbreviations used in interpretation of 1H NMR spectra are as follows: s, singlet; d, doublet; dd, doublet of doublets; t, triplet; br.t, broad triplet; q, quartet; quint, quintet; sex, sextet; sep, septet; m, multiplet; dist., distorted.
Procedure for the synthesis of 5-((2-amino-1,3-thiazol-4-yl)acetohydrazide (2)
Ethyl 2-(2-amino-1,3-thiazol-4-yl)acetate (1; 10 g, 0.054 mol) and methanol (200 mL) were taken in a 500 mL RB flask. Hydrazine hydrate (2.5 mL, 0.054 mol) was added drop wise and the mixture was refluxed for 2 h. The reaction progress was observed by TLC using n-hexane and ethyl acetate solvent system (40:60). After completion, the reaction mixture was allowed to cool at room temperature to attain white colored precipitates, which were filtered and washed with methanol to obtain purified hydrazide, 2.
Synthesis of 5-[(2-amino-1,3-thiazol-4-yl)methyl]-1,3,4-oxadiazol-2-thiol (3)
5-((2-Amino-1,3-thiazol-4-yl)acetohydrazide (2; 4 g, 0.023 mol) was dissolved in C2H5OH (70 mL) in a 250 mL RB flask at 28 °C and then solid KOH (1.34 g, 0.023 mol) was dissolved on reflux. Carbon disulphide (3.50 mL, 0.046 mol) was poured drop wise at 28 °C and then the reaction mixture was refluxed again for 5 h. Reaction progress was noted with TLC using n-hexane and ethyl acetate solvent system (7:3). After completion of reaction, excess of ethanol was evaporated and sufficient ice cold distilled water was added followed by addition of dilute HCl to adjust pH of 4-5. Light peach colored precipitates of 5-[(2-amino-1,3-thiazol-4-yl)methyl]-1,3,4-oxadiazol-2-thiol (3) were filtered and washed with distilled water.
Synthesis of 3-bromo-N-(un/substituted-phenyl)propanamides (6a-l)
The un/substituted anilines (4a-l; 0.038 mol) were suspended in 30 mL distilled water in an iodine flask (100 mL) and aqueous Na2CO3 solution (10%, 2-3 mL) was added. 3-Bromopropanoyl chloride (5; 0.038 mol) was added gradually with vigorous manual shaking. Then this mixture was set to stir on magnetic stirrer for 2-3 h. Reaction completion was monitored by TLC. On completion, the excess ice-cold distilled water (60 mL) was added and the resulting precipitates were collected through filtration, washed with distilled water, and dried to get purified electrophiles, 6a-l.
General procedure for the synthesis of 3-({5-[(2-amino-1,3-thiazol-4-yl)methyl]-1,3,4-oxadiazol-2-yl}sulfanyl)-N-(un/substituted-phenyl)propanamides (7a-l)
5-[(2-Amino-1,3-thiazol-4-yl)methyl]-1,3,4-oxadiazol-2-thiol (3; 0.1 g, 0.467 mmol) was dissolved in N,N-dimethyl formamide (DMF, 5-10 mL) in a 100 mL RB flask. Solid LiH (0.005 g) was added and the mixture was stirred for half an hour. Then, different aforementioned electrophiles, 3-bromo-N-(un/substituted-phenyl)propanamides (6a-l; 0.467 mmol), were added and the mixture was set to stirring for 3-5 h. The progress of reaction was monitored through TLC using n-hexane and ethyl acetate solvent system (80:20). On completion, excess ice-cold distilled water was added and the precipitates obtained were filtered, washed with distilled water, and dried to acquire purified products, 7a-l.
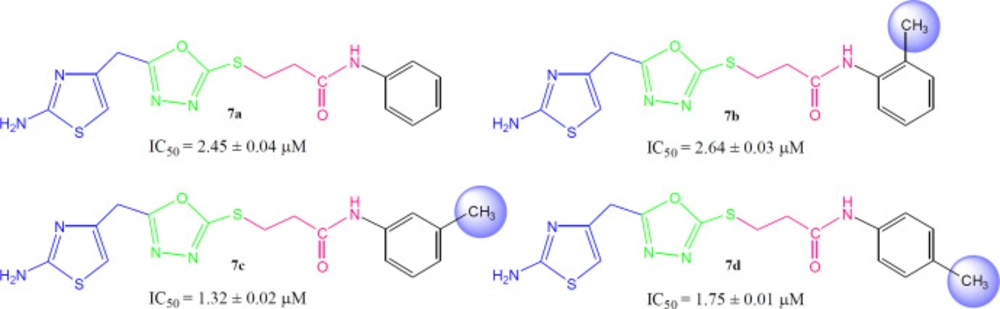
3-({5-[(2-Amino-1,3-thiazol-4-yl)methyl]-1,3,4-oxadiazol-2-yl}sulfanyl)-N-phenylpropanamide (7a)
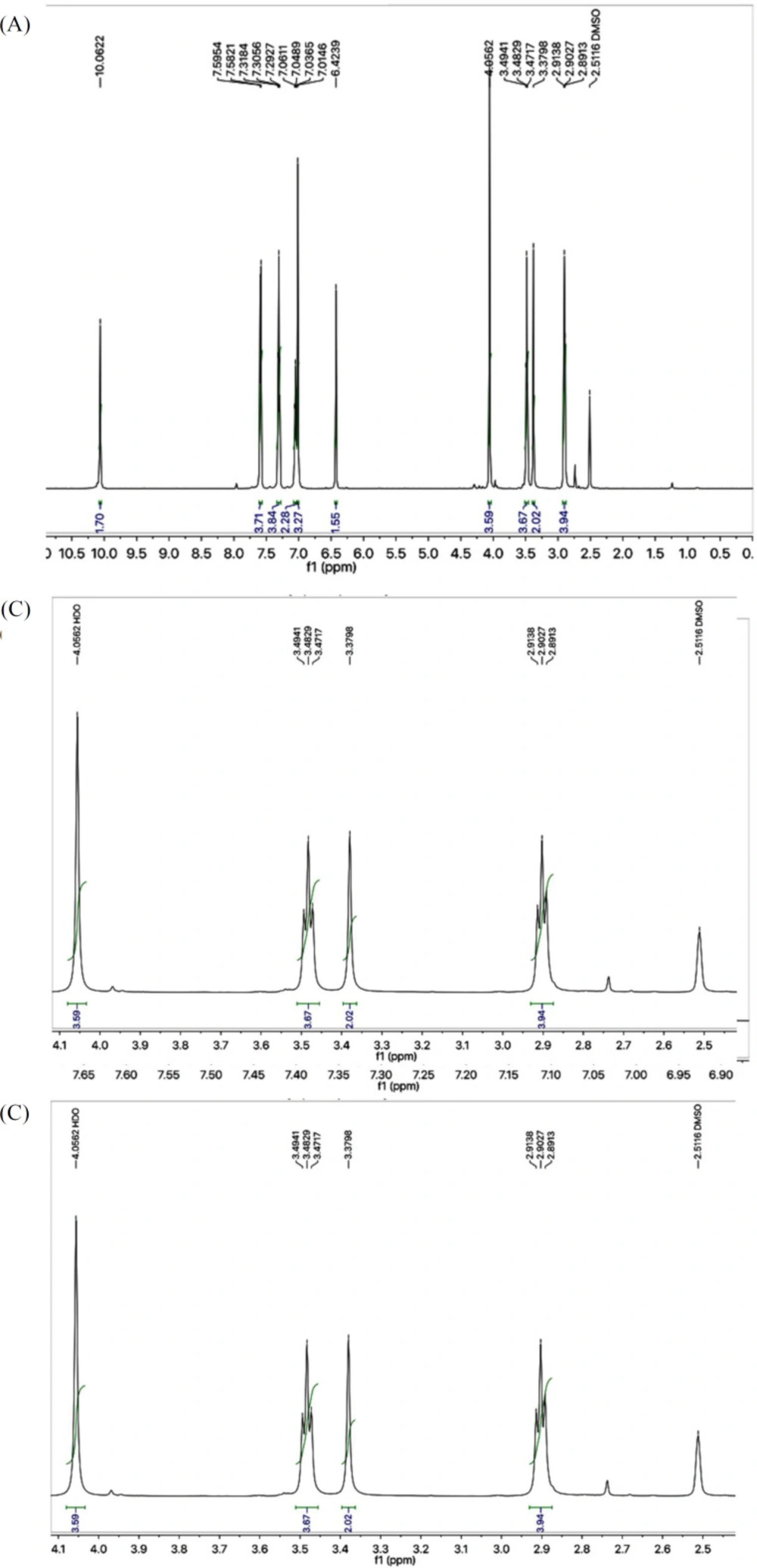
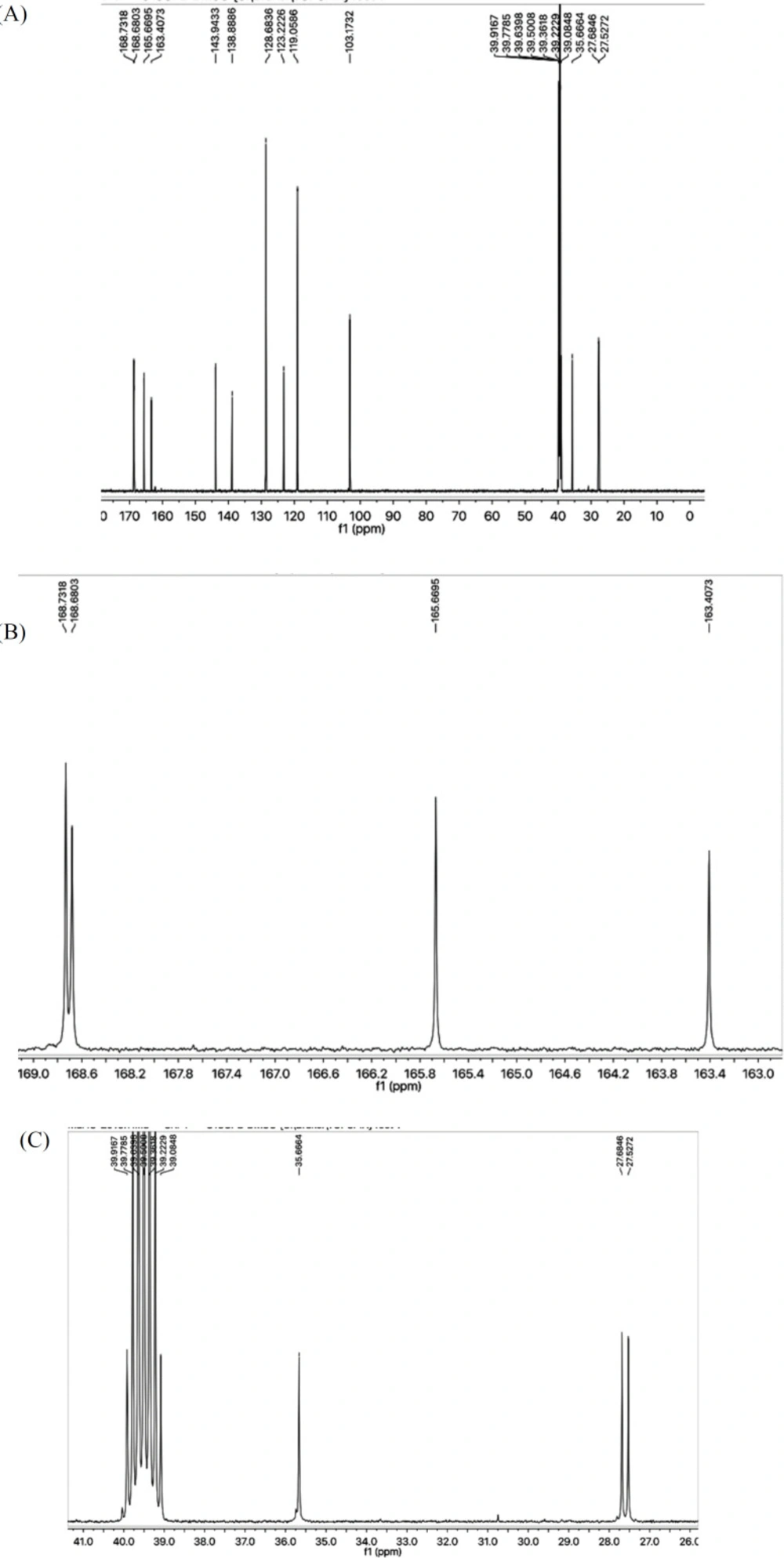
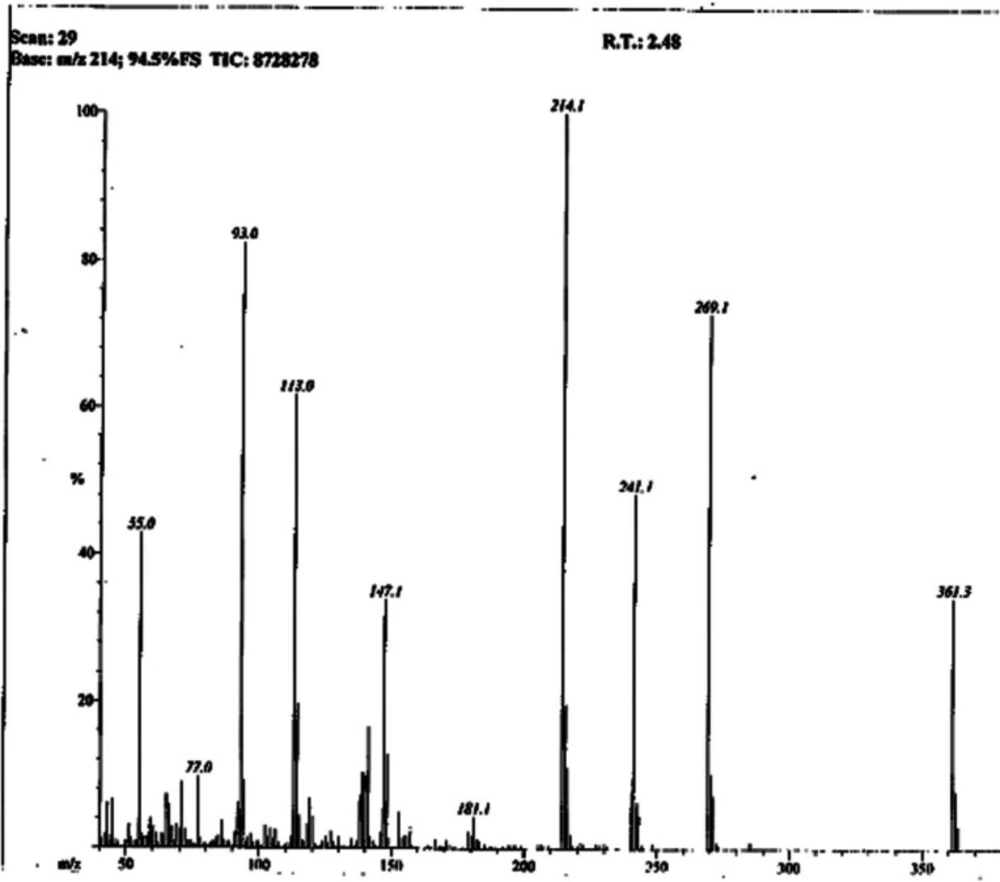
Light brown solid; Yield: 89%; m.p.:190-191 °C; Mol. Formula: C15H15N5O2S2; Mol. Mass: 361 gmol-1; IR (KBr, ʋ/cm-1): 3355 (N-H stretching), 3050 (C-H of aromatic ring), 2923 (-CH2- stretching), 1665 (C=O stretching), 1645 (C=N stretching), 1577 (C=C stretching of aromatic ring),; 1H-NMR (DMSO-d6, 600 MHz, δ/ppm): 10.06 (s, 1H, -NH-CO-1’’), 7.59 (br.d, 2H, J = 7.9 Hz, H-2’’’ and H-6’’’), 7.30 (br.t, J = 7.7 Hz, 2H, H-3’’’ and H-5’’’), 7.04 (br.t, 1H, J = 7.4, H-4’’’), 7.01 (s, 2H, -NH2), 6.42 (s, 1H, H-5), 4.05 (br.s, 2H, CH2-6), 3.48 (br.t, 2H, J = 6.7, CH2-3’’), 2.89 (br.t, 2H, J = 6.7 Hz, CH2-2’’); 13C-NMR (DMSO-d6, 150 MHz, δ/ppm): 168.73 (C-1’’), 168.68 (C-5’), 165.66 (C-2’), 163.40 (C-2), 143.94 (C-4), 138.38 (C-1’’’), 128.68 (C-3’’’ & 5’’’), 123.22 (C-4’’’), 119.05 (C-2’’’ & C-6’’’), 103.17 (C-5), 35.66 (C-6), 27.68 (C-2’’), 27.52 (C-3’’); EI-MS: m/z 361 [M]+, 269 (C9H9N2O2S2)+, 241 (C8H9N4OS2)+, 214 (C6H5N4OS2)+, 113 (C4H5N2S)+, 93 (C6H7N)+, 77 (C6H5)+, 55 (C3H3O)+.
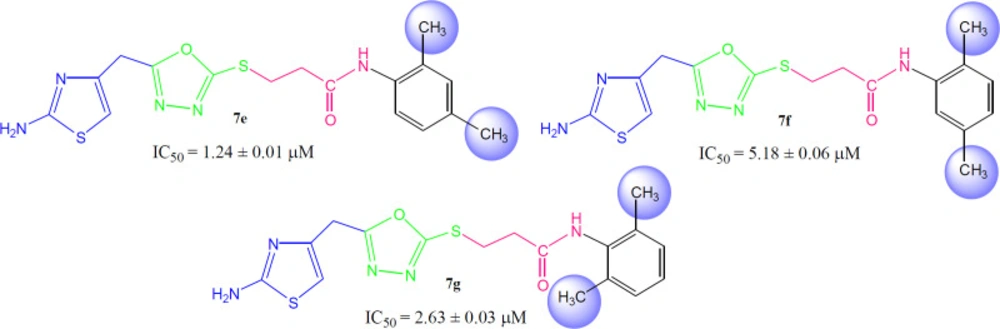
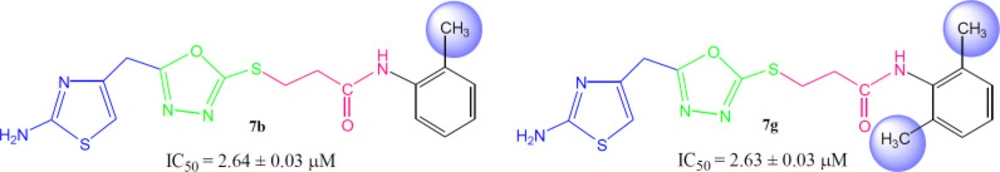
3-({5-[(2-Amino-1,3-thiazol-4-yl)methyl)-1,3,4-oxadiazol-2-yl}sulfanyl)-N-(2-methylphenyl)propanamide (7b)
Light brown solid; Yield: 85%; m.p.: 201-202 °C; Mol. Formula: C18H17N5O2S2; Mol. Mass: 375 gmol-1; IR (KBr, ʋ/cm-1): 3360 (N-H stretching), 3045 (C-H of aromatic ring), 2935 (-CH2- stretching), 1661 (C=O stretching), 1647 (C=N stretching), 1576 (C=C stretching of aromatic ring); 1H-NMR (DMSO-d6, 600 MHz, δ/ppm): 9.42 (s, 1H, -NH-CO-1’’), 7.37 (br.d, J = 7.8 Hz, 1H, H-6’’’), 7.20 (br.d, J = 7.3 Hz, 1H, H-3’’’), 7.15 (br.t, J = 7.6 Hz, 1H, H-5’’’), 7.08 (br.t, J = 7.3 Hz, 1H, H-4’’’), 7.02 (br.s, 2H, -NH2), 6.42 (s, 1H, H-5), 4.05 (br.s, 2H, CH2-6), 3.47 (br.t, J = 6.7 Hz, 2H, CH2-3’’), 2.90 (br.t, J = 6.7 Hz, 2H, CH2-2’’), 2.17 (br.s, 3H, 2’’’-CH3); 13C-NMR (DMSO-d6, 150 MHz, δ/ppm): 168.65 (C-1’’), 168.58 (C-5’), 165.55 (C-2’), 163.34 (C-2), 143.71 (C-4), 135.97 (C-1’’’), 131.62 (C-2’’’), 130.16 (C-6’’’), 125.79 (C-5’’’), 125.13 (C-3’’’), 124.94 (C-4’’’), 103.11 (C-5), 35.66 (C-6), 27.86 (C-2’’), 27.40 (C-3’’), 17.75 (2’’’-CH3); EI-MS: m/z 375 [M]+, 269 (C9H9N4O2S2)+, 241 (C8H9N4OS2)+, 214 (C6H5N4OS2)+, 195 (C10H13NOS)+, 113 (C4H5N2S)+, 106 (C7H8N) +, 55 (C3H3O)+.
3-({5-[(2-Amino-1,3-thiazol-4-yl)methyl]-1,3,4-oxadiazol-2-yl}sulfanyl)-N-(3-methylphenyl)propanamide (7c)
Light brown solid; Yield: 82%; m.p.: 171-172 °C; Mol. Formula: C16H17N5O2S2; Mol. Mass: 375 gmol-1; IR (KBr, ʋ/cm-1): 3350 (N-H stretching), 3052 (C-H of aromatic ring), 2923 (-CH2- stretching), 1670 (C=O stretching), 1641 (C=N stretching), 1576 (C=C stretching of aromatic ring); 1H-NMR (DMSO-d6, 600 MHz, δ/ppm): 9.48 (s, 1H, -NH-CO-1’’), 7.32 (br.d, J = 7.5 Hz, 1H, H-6’’’), 7.29 (br.s, 1H, H-2’’’), 7.25-7.23 (m, 2H, H-4’’’, H-5’’’), 6.99 (br.s, 2H, -NH2), 6.43 (s, 1H, H-5), 4.06 (br.s, 2H, CH2-6), 3.46 (br.t, 2H, J = 6.7 Hz, CH2-3’’), 2.91 (br.t, 2H, J = 6.7 Hz, CH2-2’’), 2.25 (br.s, 3H, 3’’’-CH3); 13C-NMR (DMSO-d6, 150 MHz, δ/ppm): 168.73 (C-1’’), 168.69 (C-5’), 165.66 (C-2’), 163.41 (C-2), 143.90 (C-4), 138.01 (C-1’’’), 137.62 (C-3’’’), 128.68 (5’’’), 123.94 (C-4’’’), 119.64 (C-6’’’), 116.28 (C-2’’’), 103.17 (C-5), 35.66 (C-6), 27.87 (C-2’’), 27.42 (C-3’’), 21.12 (3’’’-CH3); EI-MS: m/z 375 [M]+, 241 (C8H9N4OS2)+, 214 (C6H5N4OS2)+, 195 (C10H13NOS)+, 113 (C4H5N2S)+, 107 (C7H9N)+, 91 (C7H7)+, 55 (C3H3O)+.
3-({5-[(2-Amino-1,3-thiazol-4-yl)methyl]-1,3,4-oxadiazol-2-yl}sulfanyl)-N-(4-methylphenyl)propanamide (7d)
Light brown solid; Yield: 80 %; m.p.: 177-178 °C; Mol. Formula: C16H17N5O2S2; Mol. Mass: 375 gmol-1; IR (KBr, ʋ/cm-1): 3360 (N-H stretching), 3050 (C-H of aromatic ring), 2923 (-CH2- stretching), 1666 (C=O stretching), 1643 (C=N stretching), 1580 (C=C stretching of aromatic ring); 1H-NMR (DMSO-d6, 600 MHz, δ/ppm): 9.95 (s, 1H, -NH-CO-1’’), 7.45 (br.d, J = 8.4 Hz, 2H, H-2’’’ and H-6’’’), 7.11 (br.d, J = 8.3 Hz, 2H, H-3’’’ and H-5’’’), 7.00 (br.s, 2H, -NH2), 6.42 (s, 1H, H-5), 4.05 (br.s, 2H, CH2-6), 3.46 (br.t, 2H, J = 6.7, CH2-3’’), 2.86 (br.t, 2H, J = 6.7, CH2-2’’), 2.25 (s, 3H, 4’’’-CH3); 13C-NMR (DMSO-d6, 150 MHz, δ/ppm): 168.71 (C-1’’), 168.40 (C-5’), 165.66 (C-2’), 163.40 (C-2), 143.94 (C-4), 136.39 (C-1’’’), 132.10 (C-4’’’), 129.05 (C-3’’’ and C-5’’’), 119.06 (C-2’’’ and C- 6’’’), 103.15 (C-5), 35.59 (C-6), 27.71 (C-2’’), 27.51 (C-3’’), 20.39 (4’’’-CH3); EI-MS: m/z 375 [M]+, 269 (C9H9N4O2S2)+, 241 (C8H9N4OS2)+, 214 (C6H5N4OS2)+, 195 (C10H13NOS) +, 113 (C4H5N2S)+, 107 (C7H9N) +, 55 (C3H3O)+.
3-({5-[(2-Amino-1,3-thiazol-4-yl)methyl]-1,3,4-oxadiazol-2-yl}sulfanyl)-N-(2,4-dimethylphenyl)propanamide (7e)
Light brown solid; Yield: 80%; m.p.: 171-172 °C; Mol. Formula: C17H19N5O2S2; Mol. Mass: 389 gmol-1; IR (KBr, ʋ/cm-1): 3360 (N-H stretching), 3045 (C-H of aromatic ring), 2923 (-CH2- stretching), 1668 (C=O stretching), 1647 (C=N stretching), 1546 (C=C stretching of aromatic ring); 1H-NMR (DMSO-d6, 600 MHz, δ/ppm): 9.95 (s, 1H, -NH-CO-1’’), 7.23 (br.d, J = 7.5 Hz, 1H, H-6’’’), 7.04 (br.s, 1H H-3’’’), 7.01 (br.s, 2H, -NH2), 6.94 (dist.d, J = 7.6 Hz, 1H, H-5’’’), 6.42 (s, 1H, H-5), 4.05 (br.s, 2H, CH2-6), 3.48 (br.t, 2H, J = 6.7 Hz, CH2-3’’), 2.89 (br.t, 2H, J = 6.7 Hz, CH2-2’’), 2.23 (br.s, 3H, 4’’’-CH3), 2.12 (br.s, 3H, 2’’’-CH3); 13C-NMR (DMSO-d6, 150 MHz, δ/ppm): 168.72 (C-1’’), 168.68 (C-5’), 165.66 (C-2’), 163.40 (C-2), 143.90 (C-4), 134.26 (C-1’’’), 133.47 (C-2’’’), 131.65 (C-4’’’), 130.74 (C-5’’’), 126.35 (C-3’’’), 125.07 (C-6’’’), 103.17 (C-5), 35.66 (C-6), 27.68 (C-2’’), 27.52 (C-3’’), 20.42 (4’’’-CH3), 17.76 (2’’’-CH3); EI-MS: m/z 375 [M]+, 269 (C9H9N4O2S2)+, 241 (C8H9N4OS2)+, 214 (C6H5N4OS2)+, 195 (C10H13NOS)+, 121 (C8H11N)+, 106 (C8H10), 55 (C3H3O)+.
3-({5-[(2-Amino-1,3-thiazol-4-yl)methyl]-1,3,4-oxadiazol-2-yl}sulfanyl)-N-(2,5-dimethylphenyl)propanamide (7f)
Dull white solid; Yield: 89 %; m.p.: 134-135 °C; Mol. Formula: C17H19N5O2S2; Mol. Mass: 389 gmol-1; IR (KBr, ʋ/cm-1): 3345 (N-H stretching), 3050 (C-H of aromatic ring), 2923 (-CH2- stretching), 1666 (C=O stretching), 1641 (C=N stretching), 1546 (C=C stretching of aromatic ring); 1H-NMR (DMSO-d6, 600 MHz, δ/ppm): 9.40 (s, 1H, -NH-CO-1’’), 7.59 (dist.s, 1H, H-6’’’), 7.30 (br.d, J = 7.1 Hz, 1H, H-3’’’), 7.06 (br.d, J = 7.2 Hz, 1H, H-4’’’), 7.01 (br.s, 2H, -NH2), 6.42 (s, 1H, H-5), 4.05 (br.s, 2H, CH2-6), 3.48 (br.t, 2H, J = 6.7 Hz, CH2-3’’), 2.89 (br.t, 2H, J = 6.7 Hz, CH2-2’’), 2.18 (br.s, 3H, 5’’’-CH3), 2.16 (br.s, 3H, 2’’’-CH3); 13C-NMR (DMSO-d6, 150 MHz, δ/ppm): 168.73 (C-1’’), 168.68 (C-5’), 165.66 (C-2’), 163.40 (C-2), 143.90 (C-4), 137.99 (C-1’’’), 131.62 (5’’’), 130.61 (C-2’’’), 126.65 (C-3’’’), 123.23 (C-4’’’), 119.05 (C-6’’’), 103.17 (C-5), 35.66 (C-6), 27.68 (C-2’’), 27.52 (C-3’’), 19.60 (5’’’-CH3), 18.80 (2’’’-CH3); EI-MS: m/z 389 [M]+, 269 (C9H9N4O2S2)+, 241 (C8H9N4OS2)+, 214 (C6H5N4OS2)+, 175 (C11H13NO)+, 120 (C8H10N)+, 106 (C8H10), 55 (C3H3O)+.
3-({5-[(2-Amino-1,3-thiazol-4-yl)methyl]-1,3,4-oxadiazol-2-yl}sulfanyl)-N-(2,6-dimethylphenyl)propanamide (7g)
Light brown solid; Yield: 87%; m.p.: 149-150 °C; Mol. Formula: C17H19N5O2S2; Mol. Mass: 389 gmol-1; IR (KBr, ʋ/cm-1): 3360 (N-H stretching), 3020 (C-H of aromatic ring), 2923 (-CH2- stretching), 1667 (C=O stretching), 1651 (C=N stretching), 1580 (C=C stretching of aromatic ring); 1H-NMR (DMSO-d6, 600 MHz, δ/ppm): 9.37 (s, 1H, -NH-CO-1’’), 7.95 (br.s, 2H, -NH2), 7.05 (dist.d, J = 6.9 Hz, 2H, H-3’’’ and H-5’’’), 6.98 (dist.s, H-4’’’), 6.42 (s, 1H, H-5), 4.05 (br.s, 2H, CH2-6), 3.48 (br.t, 2H, J = 6.6 Hz, CH2-3’’), 2.89 (br.t, 2H, J = 6.6 Hz, CH2-2’’), 2.12 (br.s, 6H, 2’’’-CH3 and 6’’’-CH3); 13C-NMR (DMSO-d6, 150 MHz, δ/ppm): 168.72 (C-1’’), 168.29 (C-5’), 165.65 (C-2’), 163.41 (C-2), 143.94 (C-4), 135.06 (C-2’’’ and C-6’’’), 134.91 (C-1’’’), 127.62 (C-3’’’ & C-5’’’), 126.40 C-4’’’), 103.17 (C-5), 34.62 (C-6), 28.11 (C-2’’), 27.52 (C-3’’), 18.09 (2’’’-CH3 & 6’’’-CH3); EI-MS: m/z 389 [M]+, 269 (C9H9N4O2S2)+, 241 (C8H9N4OS2)+, 214 (C6H5N4OS2)+, 175 (C11H13NO)+, 121 (C8H11N)+, 106 (C8H10), 55 (C3H3O)+.
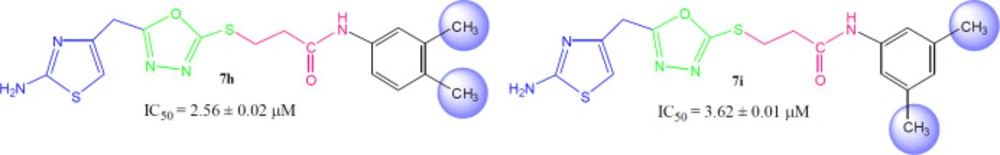
3-({5-[(2-Amino-1,3-thiazol-4-yl)methyl]-1,3,4-oxadiazol-2-yl}sulfanyl)-N-(3,4-dimethylphenyl)propanamide (7h)
Dark brown solid; Yield: 81%; m.p.: 177-178 °C; Mol. Formula: C17H19N5O2S2; Mol. Mass: 389 gmol-1; IR (KBr, ʋ/cm-1): 3350 (N-H stretching), 3052 (C-H of aromatic ring), 2923 (-CH2- stretching), 1669 (C=O stretching), 1648 (C=N stretching), 1570 (C=C stretching of aromatic ring); 1H-NMR (DMSO-d6, 600 MHz, δ/ppm): 9.96 (s, 1H, -NH-CO-1’’), 7.27 (br.d, 1H, J = 2.1 Hz, H-2’’’), 7.22 (dd, 1H, J = 2.2, 8.1 Hz, H-5’’’), 7.04 (br.d, 1H, J = 8.1 Hz, H-6’’’), 7.00 (br.s, 2H, -NH2), 6.41 (s, 1H, H-5), 4.05 (br.s, 2H, CH2-6), 3.46 (br.t, 2H, J = 6.7 Hz, CH2-3’’), 2.85 (br.t, 2H, J = 6.7 Hz, CH2-2’’), 2.20 (br.s, 3H, 4’’’-CH3), 2.16 (br.s, 3H, 3’’’-CH3); 13C-NMR (DMSO-d6, 150 MHz, δ/ppm): 168.71 (C-1’’), 168.63 (C-5’), 165.65 (C-2’), 163.39 (C-2), 143.94 (C-4), 136.70 (C-1’’’), 136.62 (C-3’’’), 131.98 (C-4’’’), 126.35 (C-5’’’), 120.49 (C-2’’’), 120.31 (C- 6’’’), 103.14 (C-5), 35.57 (C-6), 27.73 (C-2’’), 27.51 (C-3’’), 19.59 (3’’’-CH3), 18.75 (4’’’-CH3); EI-MS: m/z 389 [M]+, 269 (C9H9N4O2S2)+, 241 (C8H9N4OS2)+, 214 (C6H5N4OS2)+, 175 (C11H13NO)+, 120 (C8H10N)+, 106 (C8H10), 55 (C3H3O)+.
3-({5-[(2-Amino-1,3-thiazol-4-yl)methyl]-1,3,4-oxadiazol-2-yl}sulfanyl)-N-(3,5-dimethylphenyl)propanamide (7i)
Light brown solid; Yield: 82%; m.p.: 180-181 °C; Mol. Formula: C17H19N5O2S2; Mol. Mass: 389 gmol-1; IR (KBr, ʋ/cm-1): 3350 (N-H stretching), 3050 (C-H of aromatic ring), 2920 (-CH2- stretching), 1667 (C=O stretching), 1651 (C=N stretching), 1580 (C=C stretching of aromatic ring); 1H-NMR (DMSO-d6, 600 MHz, δ/ppm): 9.89 (s, 1H, -NH-CO-1’’), 7.21 (dist.s, 2H, H-2’’’ and H-6’’’), 7.19 (br.s, 2H, -NH2), 6.98 (br.s, H-4’’’), 6.42 (s, 1H, H-5), 4.05 (br.s, 2H, CH2-6), 3.48 (br.t, 2H, J = 6.6 Hz, CH2-3’’), 2.88 (br.t, J = 6.6 Hz, CH2-2’’), 2.12 (br.s, 6H, 3’’’-CH3 and 5’’’-CH3); 13C-NMR (DMSO-d6, 150 MHz, δ/ppm): 168.72 (C-1’’), 168.29 (C-5’), 165.65 (C-2’), 163.41 (C-2), 143.94 (C-4), 137.65 (C-1’’’), 135.06 (C-3’’’ and C-5’’’), 127.62 (C-4’’’), 124.34 (C-2’’’ and C-6’’’), 103.17 (C-5), 35.62 (C-6), 28.11 (C-2’’), 27.52 (C-3’’), 21.06 (3’’’-CH3 and 5’’’-CH3); EI-MS: m/z 389 [M]+, 269 (C9H9N4O2S2)+, 241 (C8H9N4OS2)+, 214 (C6H5N4OS2)+, 175 (C11H13NO)+, 121 (C8H11N)+, 106 (C8H10), 55 (C3H3O)+.
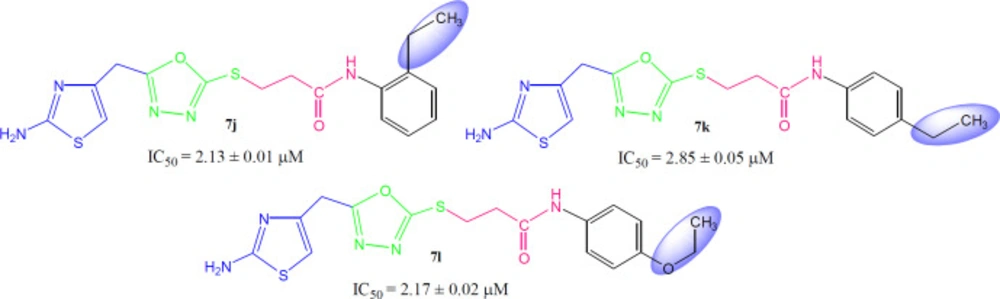
3-({5-[(2-Amino-1,3-thiazol-4-yl)methyl]-1,3,4-oxadiazol-2-yl}sulfanyl)-N-(2-ethylphenyl)propanamide (7j)
Light brown solid; Yield: 91%; m.p.: 198-199 °C; Mol. Formula: C17H19N5O2S2; Mol. Mass: 389 gmol-1; IR (KBr, ʋ/cm-1): 3350 (N-H stretching), 3075 (C-H of aromatic ring), 2930 (-CH2- stretching), 1667 (C=O stretching), 1650 (C=N stretching), 1576 (C=C stretching of aromatic ring); 1H-NMR (DMSO-d6, 600 MHz, δ/ppm): δ 9.42 (s, 1H, -NH-CO-1’’), 7.32 (dist.dd, 1H, J = 1.3, 7.2 Hz, H-6’’’), 7.22 (dd, J = 1.5, 7.1 Hz, 1H, H-3’’’), 7.17-7.12 (m, 2H, H-4’’’ and H-5’’’), 6.99 (br.s, 2H, -NH2), 6.41 (s, 1H, H-5), 4.05 (br.s, 2H, CH2-6), 3.47 (br.t, J = 6.7 Hz, 2H, CH2-3’’), 2.90 (br.t, J = 6.7 Hz, CH2-2’’), 2.56 (q, J = 7.6 Hz, 2H, 2’’’-CH2-CH3), 1.09 (t, 3H, J = 7.6 Hz, 2’’’-CH2-CH3); 13C-NMR (DMSO-d6, 150 MHz, δ/ppm): 168.99 (C-5’), 168.71 (C-1’’), 165.65 (C-2’), 163.42 (C-2), 143.94 (C-4), 137.91 (C-1’’’), 135.30 (C-2’’’), 128.48 (C-3’’’), 126.03 (C-5’’’), 125.84 (C-4’’’), 125.71 (C-6’’’), 103.15 (C-5), 35.01 (C-6), 27.97 (C-2’’), 27.52 (C-3’’), 23.72 (2’’’-CH2-CH3), 14.21 (2’’’-CH2-CH3); EI-MS: m/z 389 [M]+, 269 (C9H9N4O2S2)+, 241 (C8H9N4OS2)+, 214 (C6H5N4OS2)+, 175 (C11H13NO)+, 121 (C8H11N)+, 106 (C8H10), 55 (C3H3O)+.
3-({5-[(2-amino-1,3-thiazol-4-yl)methyl]-1,3,4-oxadiazol-2-yl}sulfanyl)-N-(4-ethylphenyl)propanamide (7k)
Light brown solid; Yield: 84 %; m.p.: 193-194 °C; Mol. Formula: C17H19N5O2S2; Mol. Mass: 389 gmol-1; IR (KBr, ʋ/cm-1): 3345 (N-H stretching), 3062 (C-H of aromatic ring), 2923 (-CH2- stretching), 1663 (C=O stretching), 1649 (C=N stretching), 1576 (C=C stretching of aromatic ring); 1H-NMR (DMSO-d6, 600 MHz, δ/ppm): 9.96 (s, 1H, -NH-CO-1’’), 7.47 (br.d, J = 8.5 Hz, 2H, H-2’’’ and H-6’’’), 7.13 (br.d, J = 8.5 Hz, 2H, H-3’’’ and H-5’’’), 6.99 (br.s, 2H, -NH2), 6.41 (s, 1H, H-5), 4.04 (br.s, 2H, CH2-6), 3.45 (br.t, 2H, J = 6.7, CH2-3’’), 2.86 (br.t, 2H, J = 6.7, CH2-2’’), 2.54 (q, 2H, J = 7.5 Hz, 4’’’-CH2-CH3), 1.14 (t, 3H, J = 7.5 Hz, 4’’’-CH2-CH3); 13C-NMR (DMSO-d6, 150 MHz, δ/ppm): 168.71 (C-1’’), 168.41 (C-5’), 165.66 (C-2’), 163.40 (C-2), 143.94 (C-4), 138.59 (C-1’’’), 136.58 (C-4’’’), 127.86 (C-3’’’ and C-5’’’), 119.14 (C-2’’’ and C-6’’’), 103.15 (C-5), 35.59 (C-6), 27.72 (C-2’’), 27.54 (4’’’-CH2-CH3), 27.51 (C-3’’), 15.64 (4’’’-CH2-CH3); EI-MS: m/z 389 [M]+, 269 (C9H9N4O2S2)+, 241 (C8H9N4OS2)+, 214 (C6H5N4OS2)+, 175 (C11H13NO)+, 121 (C8H11N)+, 106 (C8H10), 55 (C3H3O)+.
3-({5-[(2-Amino-1,3-thiazol-4-yl)methyl]-1,3,4-oxadiazol-2-yl}sulfanyl)-N-(4-ethoxyphenyl)propanamide (7l)
Light brown solid; Yield: 80%; m.p.: 177-178 °C; Mol. Formula: C16H17N5O2S2; Mol. Mass: 375 gmol-1; IR (KBr, ʋ/cm-1): 3360 (N-H stretching), 3050 (C-H of aromatic ring), 2923 (-CH2- stretching), 1663 (C=O stretching), 1651 (C=N stretching), 1580 (C=C stretching of aromatic ring); 1H-NMR (DMSO-d6, 600 MHz, δ/ppm): 9.88 (s, 1H, -NH-CO-1’’), 7.46 (br.d, J = 9.0 Hz, 2H, H-2’’’ and H-6’’’), 6.99 (br.s, 2H, -NH2), 6.85 (br.d, J = 9.0 Hz, 2H, H-3’’’ and H-5’’’), 6.41 (s, 1H, H-5), 4.04 (br.s, 2H, CH2-6), 3.97 (q, J = 6.9 Hz, 2H, 4’’’-OCH2-CH3), 3.45 (br.t, 2H, J = 6.7 Hz, CH2-3’’), 2.84 (br.t, 2H, J = 6.7 Hz, CH2-2’’), 1.29 (t, J = 6.9 Hz, 3H, 4’’’-OCH2-CH3); 13C-NMR (DMSO-d6, 150 MHz, δ/ppm): 168.64 (C-1’’), 168.00 (C-5’), 165.58 (C-2’), 163.33 (C-2), 154.34 (C-4’’’), 143.87 (C-4), 131.89 (C-1’’’), 120.50 (C-2’’’ and C-6’’’), 114.27 (C-3’’’ and C-5’’’), 103.07 (C-5), 62.96 (4’’’-OCH2-CH3) 35.42 (C-6), 27.71 (C-2’’), 27.44 (C-3’’), 14.57 (4’’’-OCH2-CH3); EI-MS: m/z 375 [M]+, 269 (C9H9N4O2S2)+, 241 (C8H9N4OS2)+, 214 (C6H5N4OS2)+, 136 (C8H10NO)+, 121 (C8H9O), 55 (C3H3O)+.
Urease inhibition assay
This enzyme assay is the customized form of the commonly known Berthelot assay (
23,
24). The assay mixture of 85 µL is prepared containing 10 µL of phosphate buffer of pH 7.0 (in each well in the 96-well plate), 10 µL of sample solution and 25 µL of enzyme solution (0.135 units). The contents were pre-incubated at 37 ºC for 5 min. 40 µL of urea stock solution (20 mM) was added to each well with incubation for 10 min at 37 ºC. It is followed by the addition of 115 µL phenol hypochlorite reagents (freshly prepared by mixing 45 µL phenol with 70 µL of alkali) per well. For color development, incubation was carried out for further 10 min at 37 ºC. The absorbance was measured at 625 nm. The percentage enzyme inhibition and IC
50 values were calculated using the following formula:
Where, Control is the total enzyme activity without inhibitor and Test is the activity in the presence of test compound. IC50 values were calculated using the EZ–Fit Enzyme kinetics software (Perrella Scientific Inc. Amherst, US).
Statistical analysis
Statistical analysis was performed by Microsoft Excel 2010 for all the thrice measured values and the results are presented as mean ± SEM.
Hemolytic activity
Bovine blood samples was collected in EDTA, that was diluted with saline (0.9% NaCl), and centrifuge at 1000 ×g for 10 min. The erythrocytes separated diluted in phosphate buffer saline of pH 7.4 and a suspension was made. Add 20 µL of synthetic compounds solution (10 mg/mL) in 180 µL of RBCs suspension and incubate for 30 min at room temperature. PBS was used as negative control and Triton 100-X was taken as positive control (
25,
26). The percentage of hemolysis was taken as by using Equation:
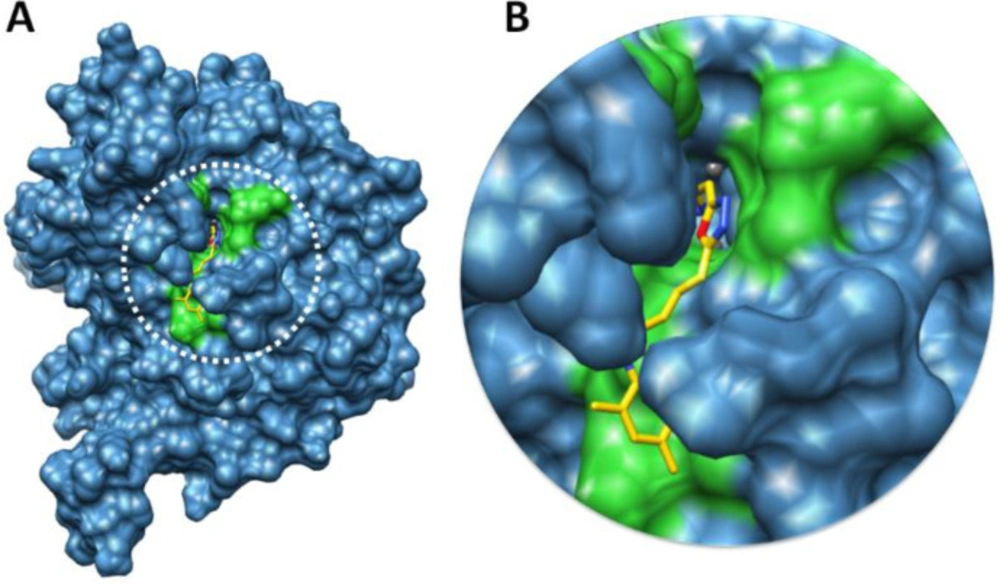
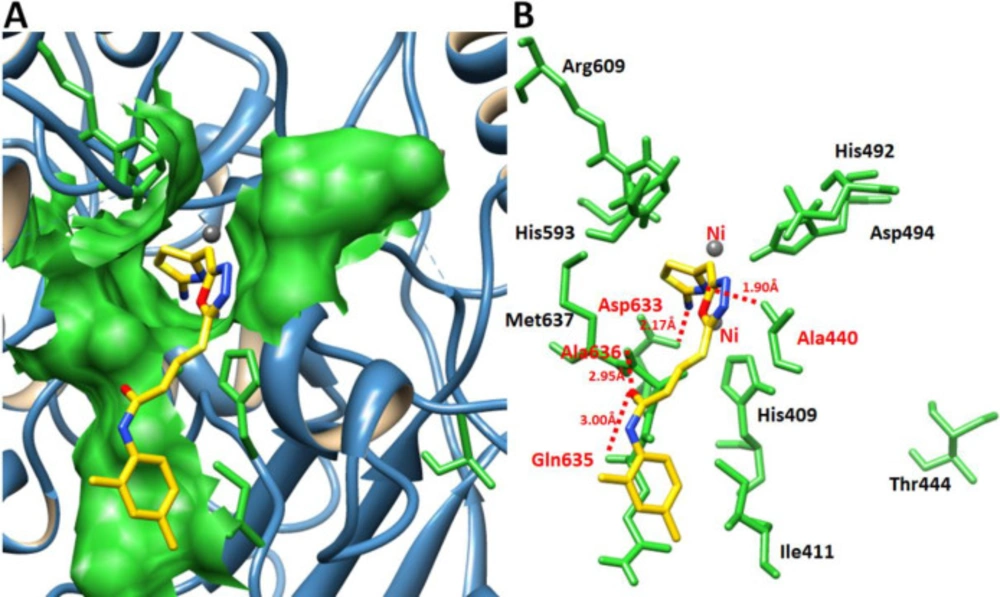
Molecular docking
Initially, the synthesized chemical ligands (
7a-l) were drawn in ACD/ChemSketch tool and retrieved in mol format. Furthermore, UCSF Chimera 1.10.1 tool was employed for energy minimization having default parameters such as steepest descent steps 100 with step size 0.02 (Å), conjugate gradient steps 100 with step size 0.02 (Å) and update interval was fixed at 10. Finally, Gasteiger charges were assigned in ligands using Dock Prep to obtain the good structure conformation. Molecular docking experiment was employed on
7a-l, against Jack bean urease by using virtual screening tool PyRx with VINA Wizard approach (
27). The grid box parameters values in VINA search space (X = 11.06, Y = -54.61 and Z = -27.12) were adjusted with default exhaustiveness value = 8 to maximize the binding conformational analysis. We have adjusted sufficient grid box size on biding pocket residues to allow the ligand to move freely in the search space. The generated docked complexes were evaluated on the basis of lowest binding energy (kcal/mol) values and binding interaction pattern between ligands and receptor. The graphical depictions of all the docked complexes were accomplished by UCSF Chimera 1.10.1 (
28) and Discovery Studio (2.1.0), respectively.

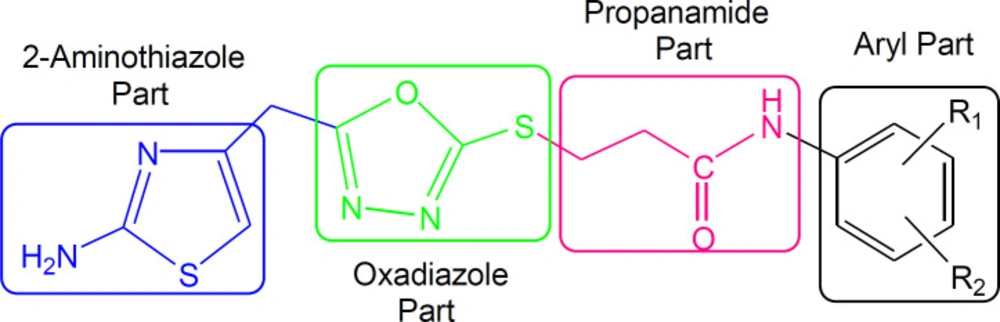
![Outline for the synthesis of 3-({5-[(2-amino-1,3-thiazol-4-yl)methyl]-1,3,4-oxadiazol-2-yl}sulfanyl)-<i>N</i>-(un/substituted-phenyl)propanamides. Reagents and Conditions: (I) MeOH/N<sub>2</sub>H<sub>4</sub><b>·</b>H<sub>2</sub>O/refluxing for 2 h. (II) EtOH/CS<sub>2</sub>/KOH/refluxing for 5 h. (III) Aq. 10% Na<sub>2</sub>CO<sub>3</sub> soln./vigorous manual shaking and stirring at RT for 2-3 h. (IV) DMF/LiH/stirring for 3-5](https://brieflands.com/journals/ijpr/articles/124559/figures/ijpr-19-487-g012-preview.webp)
