Chemicals
Polymyxin B sulfate salt, Ampicillin, Tetra-cycline, Acrylamide/bisacrylamide solution (30%), IPTG and Protein ladder were supplied from Sigma (USA). Other chemicals were purchased from Merck Chemicals (Germany). SYBR green (Safe stain) and 50 bp DNA ladder were supplied from Sinaclon (Iran). NdeI was purchased from Takara (Japan) and EcoRI from Thermo Scientific (USA).
Peptide design as a novel AMP
To design a new AMP, Magainin II was considered as the template AMP with promising activity against a broad microbial agent. Peptide sequences of magainin II and pexiganan was extracted from the antimicrobial peptide Database (APD; http://aps.unmc.edu/AP) (
22). Magainin II sequence is GIGKFLHSAKKFGKAFVGEIMNS. The peptide length was shortened and then, Lys, His, and Ser residues from magainin II sequence were substituted with Arg and also, hydrophobic Phe with Trp. Various peptide sequences were designed and analyzed by dPABBs (Design Peptides against Bacterial Biofilms) web server (http://ab-openlab.csir.res.in/abp/antibiofilm/index.php) (
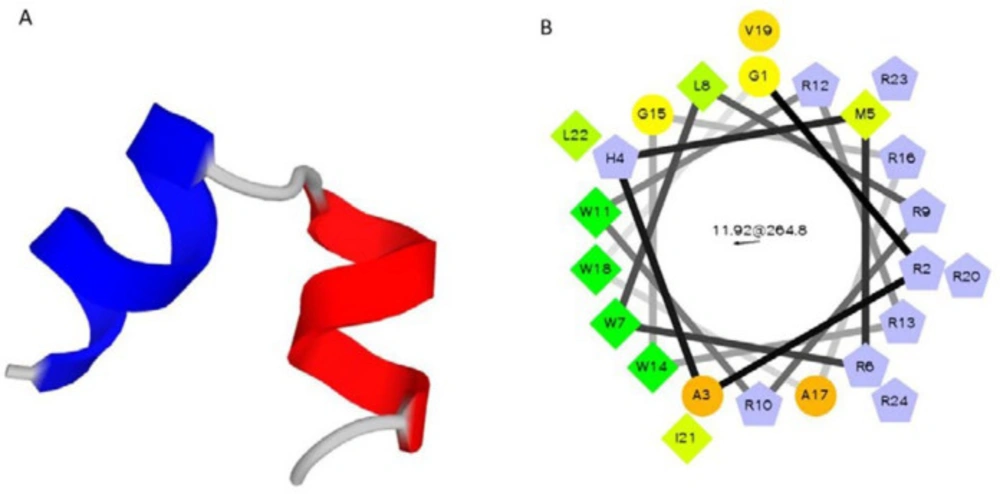
23). The best designed peptide (P19) had 19 amino acid residues. Then, according to the structure of the vector pTYB21, some modifications were applied in to the designed AMP (P19) and five amino acid residues (GRAHM) were added to the N-terminal of P19, which resulted in 24 amino acid sequence of P24. Physicochemical characteristics of the designed peptides were predicted by PepFold (http://bioserv.rpbs.univ-paris-diderot.fr/services/PEP-FOLD/) and Helical Wheel draw program from Raphael Zidovetzki’s lab (http://rzlab.ucr.edu/scripts/).
Peptide and DNA sequences
Pexiganan (GIGKFLKKAKKFGKAFVK-ILKK-NH
2) was chemically synthesized by Dr. Kobarfard group (School of Pharmacy, Shahid Beheshti University of Medical Sciences, Tehran, Iran). Vector pTYB21 (cat# N6709S,
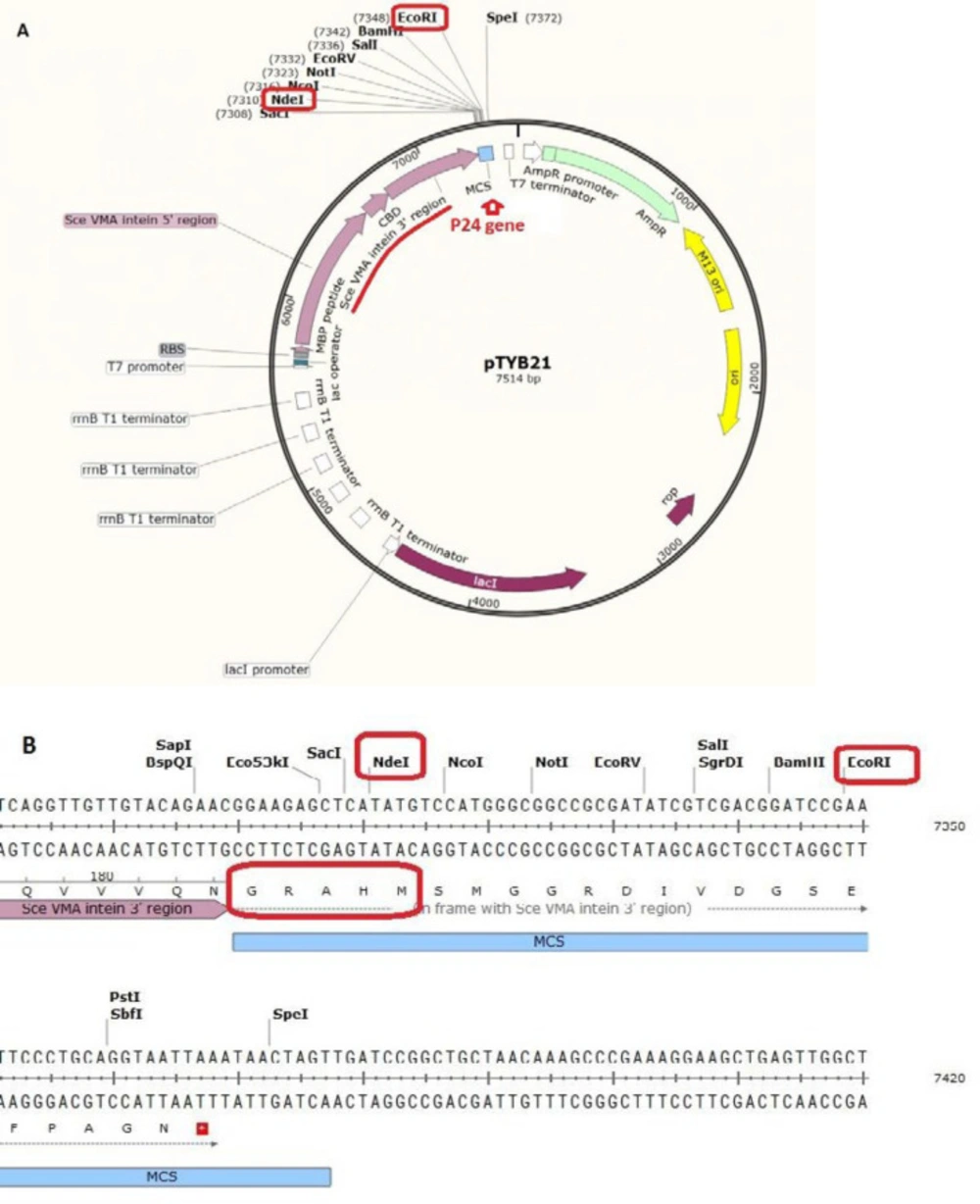
Figure 1) was supplied from New England Biolab company (UK). The vector contains chitin binding domain and Sec VMA intein sequence upstream of the multiple cloning site (MCS). pTYB21 had ampicillin resistance gene as the selection marker. Universal primers of this vector were synthesized by Sinaclon Company (Iran). After designing the peptide, the P24 AMP gene was reverse-translated from amino acid sequence using the EXPASY bioinformatics tool (Reverse translate tool) and the codons were optimized according to the
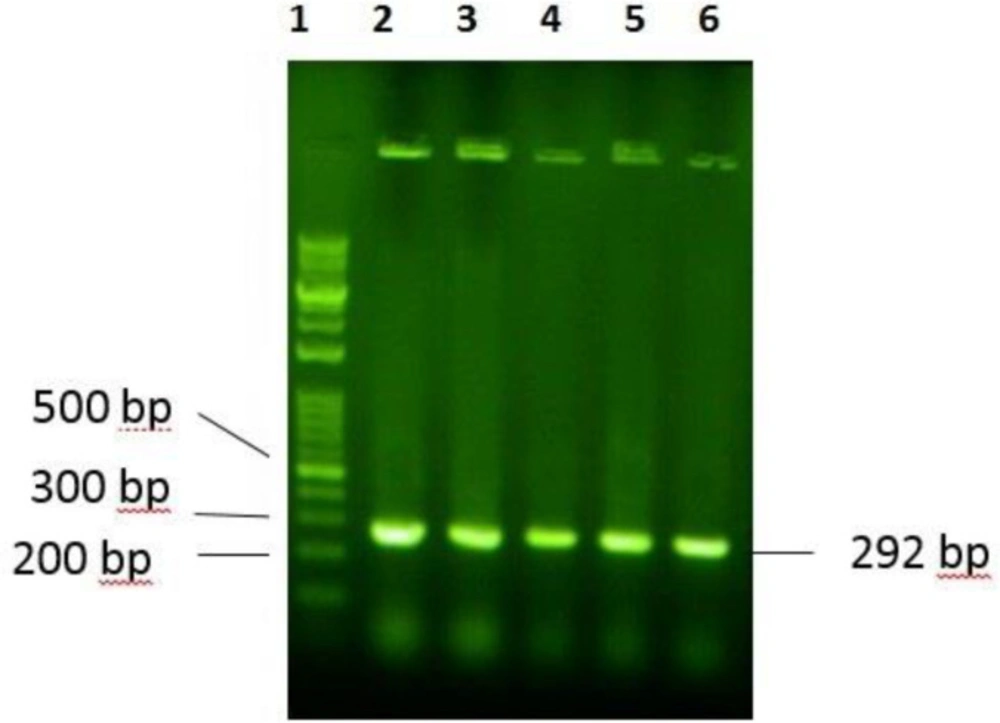
E. coli expression system. Gene plus (5´-TTTTTTT CAT ATG CGC TGG TTA CGC CGT TGG CGT CGC TGG GGC CGC GCC TGG GTC CGT ATT CTT CGC CGC TAA GAA TTC TTTTTTT- 3´) and gene minus (5´- AAAAAAA GAA TTC TTA GCG GCG AAG AAT ACG GAC CCA GGC GCG GCC CCA GCG ACG CCA ACG GCG TAA CCA GCG CAT ATG AAAAAAA- 3´) were synthesized by Bioneer (Korea). The P24 gene was designed with NdeI restriction site at 5’ end and EcoRI restriction site at the 3´end of the gene for directed cloning into the pTYB21. A stop codon was designed at the 3´end of the gene.
Cloning and transformation of AMP gene-containing construct
The P24 gene was digested with NdeI at 5’ end and EcoRI at the 3´end of the gene for site-directed cloning into the pTYB21. After confirmation of the desired recombination by polymerase chain reaction (PCR) and electrophoresis on agarose gel, the recombinant vector was cloned into the E. coli TOP10F’ that had an endogenous plasmid with tetracycline resistance gene as the selection marker). The positive clone, which grow on ampicillin and tetracycline supplemented LB-agar culture plate, was selected and the recombinant plasmid was extracted and transformed into the E. coli BL21 for protein expression using calcium chloride heat-shock method.
The fusion gene composed of the designed peptide and the intein-chitin binding domain is under the control of T7 promoter, which can be readily induced by isopropyl b-D-thiogalactoside (IPTG). From the overnight culture of
E. coli BL21, cell harboring recombinant pTYB 21 constructs was sub-cultured (1:100) in fresh LB broth medium supplemented with 0.1 mg mL
-1 ampicillin and grown at 37 °C under agitation. Expression of the fusion protein was induced by the addition of IPTG to a final concentration of 400 µmol L
-1 (when the OD
600nm of culture reached to 0.8). After induction, the cultures were incubated at 37 °C. To find the best time of cell harvesting, the 1 mL sample of culture was harvested every hour until 5 h (
19).
CBD-AMP fusion protein production in E. coli
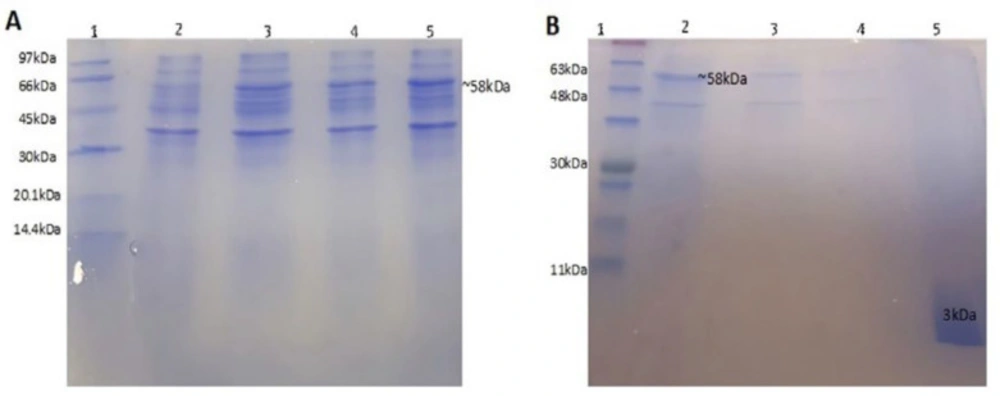
A suspension (500 mL) of recombinant
E. coli BL21 in LB broth medium was cultured at 37 °C. Four hours after IPTG induction, the cells were harvested by centrifugation at 4500 ×g, at 4 °C for 15 min and washed three times with PBS. The supernatant was removed and evaluated by SDS-PAGE 15%. The bacterial pellet was re-suspended in buffer-1 (20 mmol L
-1 Tris–HCl, pH 8.0, 0.1 mmol L
-1 EDTA, 0.5 mol L
-1 NaCl, 20 µmol L
-1 PMSF) and disrupted by sonication (200 W power for 25 min in 30 sec intervals of off/on program) (
19). Lysis mixture was centrifuged at 15000 ×g, at 4 °C for 30 min. The release of protein was monitored using the Bradford assay. The pellet and supernatant were separated and checked for the presence of the fusion protein by SDS-PAGE 15%.
Purification of the desired AMP from inclusion bodies
Pellet of inclusion bodies was solubilized in buffer-2 (urea 8M in column buffer). The mixture was vortex and centrifuged at 12000 ×g for 20 min at 4 °C. The supernatant, containing fusion protein (56 kDa), was dialyzed against buffer-1 with gradient reduction of urea concentration using 12 kDa dialysis tube (#D0405, Sigma, USA) at 37 °C to refold the protein. The resulted solution was loaded onto a 10 mL chitin column and washed with 10-bed volumes of buffer-1, then, the column was flushed quickly with 3-bed volumes of buffer-3 (elution buffer: 20 mmol L-1 Tris–HCl buffer (pH 8.0), 0.1 mmol L-1 EDTA and 0.5 mol L-1 NaCl containing 50 mmol L-1 DTT). The column was incubated at room temperature for 40 h to induce self-cleavage of the fusion protein. After that, the protein was eluted from the column by 2-bed volumes of elution buffer. The cleavage efficiency was determined using SDS-PAGE 15%.
Antimicrobial activity assay
Antibacterial activity assay was performed by microdilution method (24). Briefly, 0.1 mL of Muller-Hinton broth medium, containing CaCl2 and MgCl2 (final concentration of calcium 20 µg mL-1 and magnesium 10 µg mL-1), was added to each well of a 96-well microtiter plate. The primary stock of P24 peptide was provided to 10 mg mL-1 concentration. Serial dilution of the P24 peptide and pexiganan in the range of 4-512 µg mL-1 was prepared in separate wells in triple sets. 0.01 mL of the 1:20 dilution of 0.5 McFarland suspension (1 × 108 CFU mL-1), which contains 5 × 106 CFU mL-1 was added to each well. Bacterial suspensions included Pseudomonas aeruginosa (P. aeruginosa), Staphylococcus aureus (S. aureus), Enterococcus faecalis (E. faecalis), Escherichia coli (E. coli), Klebsiella pneumoniae (K. pneumoniae). The plates were incubated at 35 ± 2 °C and ambient air for 24 h before reading the MICs.
Hemolytic activity assay
Hemolytic activity was assayed as described by Jang
et al. (
25). Briefly, 3 mL of freshly prepared human red blood cells (RBCs) was washed with isotonic PBS (pH 7.4), until the color of the supernatant became clear. The washed RBCs were then suspended in the final volume of 20 mL using BPS, pH 7.4. Ten microliters of peptide samples, serially diluted in PBS (final concentrations ranging from 0.15 to 150 µg mL
-1), was added to 190 mL of the cell suspension in microfuge tubes. Following gentle mixing, the tubes were incubated at 37 °C for 30 min and then, centrifuged at 4000 ×g for 5 min. Finally, 100 mL of supernatant was diluted to 1 mL with PBS and then, absorbance at 567 nm was measured for monitoring the release of hemoglobin. The release of hemoglobin indicated RBC membrane damage. Negative control with no hemolysis and the positive control with 100% hemolysis were determined in PBS and 0.2% Triton X-100, respectively. The percentage of hemolysis was calculated using the following Equation:
, where A
s is the absorbance of the sample, A100 is the absorbance of completely lysed RBCs in 0.2% Triton X-100 and A
0 is the absorbance in the complete absence of haemolysis.