General methods
All the chemicals and solvents were purchased from Merck (Darmstadt, Germany). The melting points of the compounds were measured on 9100 Electrothermal melting point apparatus. Elemental analyses were performed by a Costech ECS 4010 CHNS analyzer. The 1H-NMR spectra were recorded on a 400 MHz Bruker spectrometer. IR spectra were recorded on Perkin Elmer IR spectrophotometer as potassium bromide discs. GC–MS analysis were carried out by using a 7000 Agilent triple Quadrupole MS system coupled with a 7890A GC, equipped with a split/splitless injection port, an autosampler model Agilent 7693, and electronic ionization. A HP-5MS 5% Phenyl Methyl Silox, Agilent 19091s-433 capillary column was used (30 m × 0.25 mm I.D. and 0.25 μm film thickness). Helium gas with a purity of 99.99% and a flow rate of 1 mL/min was used as the carrier gas.
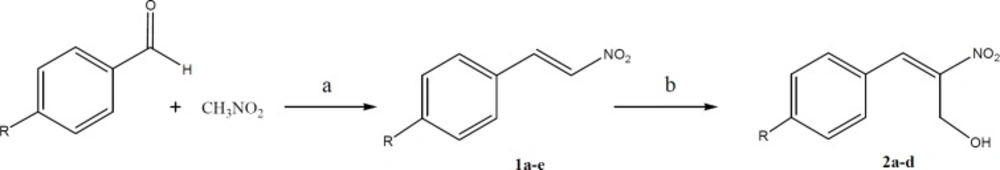
General procedure for the synthesis of para-substituted nitrostyrene (1a-e)
Nitromethane (0.1 moles) and the appropriate aldehyde (0.1 moles) were dissolved in 20 mL of methanol in a beaker that was kept cool by a mixture of ice and salt. A cold solution of sodium hydroxide (10 mL, 0.2 M) was added from an addition funnel to the stirring solution of nitromethane and benzaldehyde at a rate that the temperature was kept below 15 °C. A precipitate was formed and got thick during the addition of sodium hydroxide solution. After fifteen minutes remaining in the lab, about 60 mL of ice water was added to the mixture and the pasty mass was converted to a clear solution. The reaction mixture was added dropwise to a solution of HCl (20 mL of concentrated HCl diluted to 50 mL with water). A solid crystalline mass was formed and separated by decantation. The residue was filtered and washed with water and recrystallized from ethanol (
15).
(2-nitrovinyl)benzene (1a): Yellow solid. Yield: 83%. mp 57-58 °C. IR (KBr): 1635, 1523, 1348, 973, 778. GC-MS (m/z): 149.1 [M]+. 1H-NMR (400 MHz, CDCl3): 7.43-7.56 (5H, m, CHPh), 7.59 (1H; d, J = 13.7 Hz; =CHα), 8.01 (1H; d, J = 13.7 Hz; =CHβ). Anal. Calcd. For C8H7NO2 (149.15): C, 64.42; H, 4.73; N, 9.39; O, 21.45. Found: C, 64.37; H, 4.69; N, 9.41; O, 21.38.
1-chloro-4-(2-nitrovinyl)benzene (1b): Crystalline (needle shape), light yellowish solid. Yield: 46%. mp 111-113 °C. IR (KBr): 1627, 1587, 1525, 1343, 979. GC-MS (m/z): 183.0 [M]+. 1H-NMR (400 MHz, CDCl3): 7.44 (2H; d, J = 8.6 Hz; CHAr-O-Cl), 7.49 (2H; d, J = 8.6 Hz; CHAr-m-Cl), 7.56 (1H; d, J = 13.7 Hz; =CHα), 7.96 (1H; d, J = 13.7 Hz; =CHβ). Anal. Calcd. For C8H6ClNO2 (183.59): C, 52.34; H, 3.29; Cl, 19.31; N, 7.63; O, 17.43. Found: C, 52.32; H, 3.30; Cl, 19.30; N, 7.63; O, 17.41.
1-methyl-4-(2-nitrovinyl)benzene (1c): Yellow solid. Yield: 57%. mp 105-106 °C; IR (KBr): 1628, 1514, 1345, 967, 815. GC-MS (m/z): 163.1 [M]+. 1H-NMR (400 MHz, CDCl3): 2.41 (3H, s, Me), 7.26 (2H; d, J = 8Hz; CHAr-o-Me), 7.45 (2H; d, J = 8Hz; CHAr-m-Me), 7.57 (1H; d, J = 13.6 Hz; =CHα ), 7.99 (1H; d, J = 13.6 Hz; =CHβ). Anal. Calcd. For C9H9NO2 (163.17): C, 66.25; H, 5.56; N, 8.58; O, 19.61. Found: C, 66.22; H, 5.57; N, 8.56; O, 19.60.
The synthetic pathway for preparation of para-substituted nitrostyrenes (1a-e) and 2-nitro-3-arylprop-2-en-1-ols (2a-d). Reagents and conditions: a) NaOH, temp. < 15 °C; b) Imidazole, anthranilic acid, formaldehyde solution, r. t
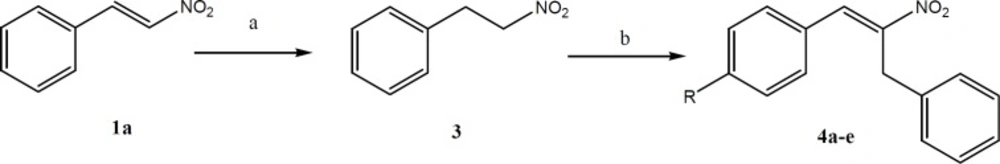
The synthetic pathway for preparation of aryl-2-nitroallylbenzene (4a-e). Reagents and conditions: a) Silica gel, NaBH4, 2-propanol and chloroform; b) Dimethylamine hydrochloride, potassium fluoride, toluene.
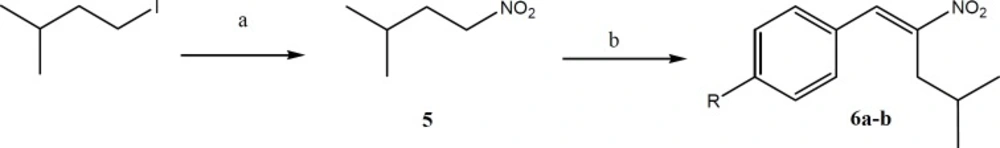
The synthetic pathway for preparation of Aryl-4-methyl-2-nitropent-1-ene (6a-b). Reagents and conditions: a) Isopentyl iodide, sodium nitrite, DMF, 0 °C; b) n-BuNH2, methanol and acetic acid, 0 °C, under argon.
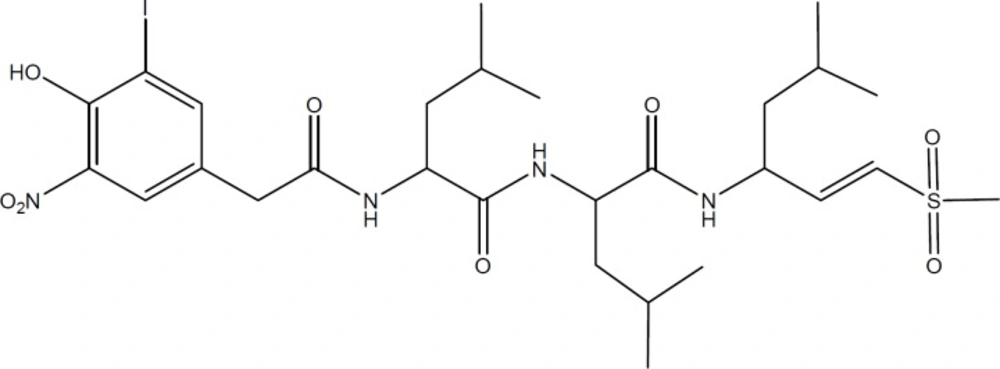
Chemical structure of NIP-Leu3-Vinyl sulfone as a representative of vinyl sulfones
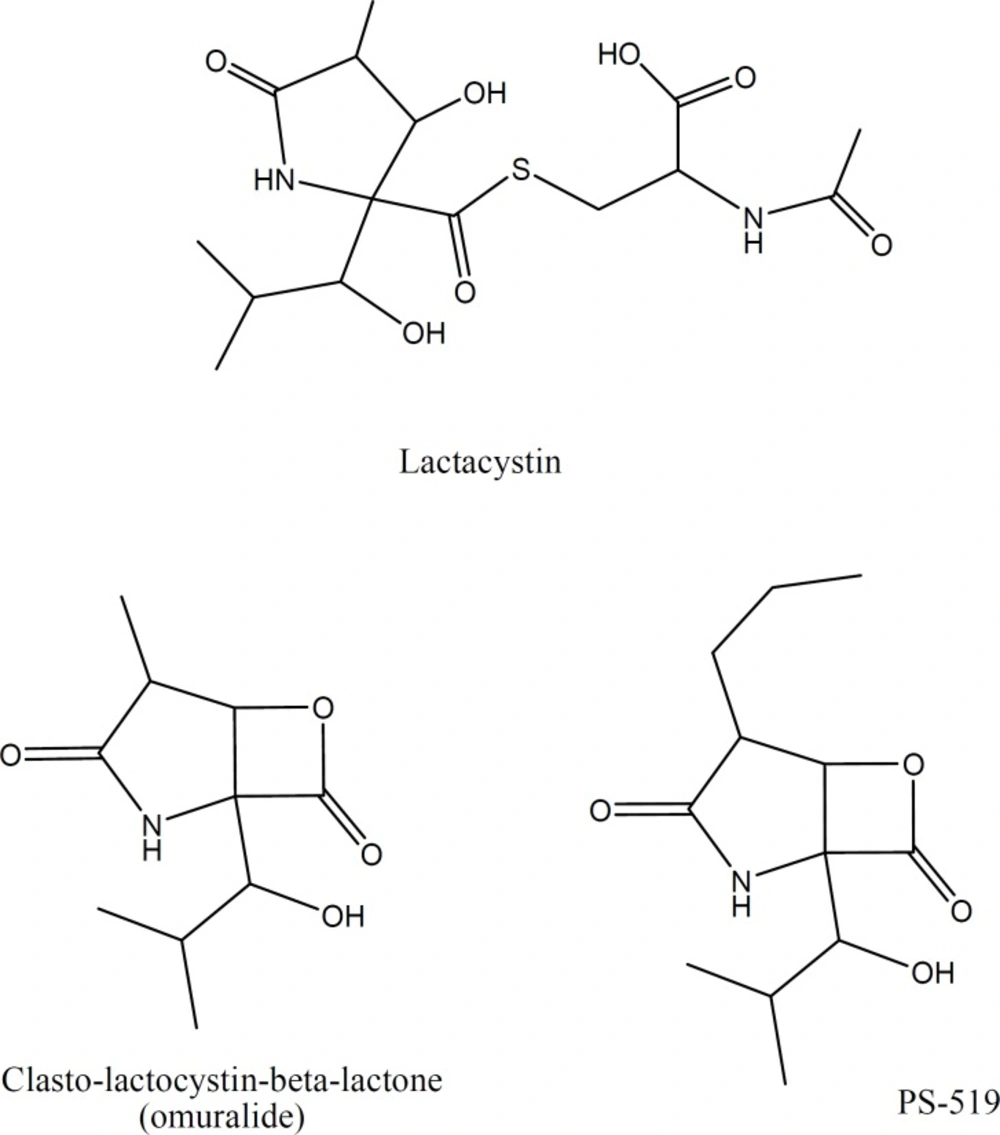
Chemical structure of Lactacystin and its derivatives
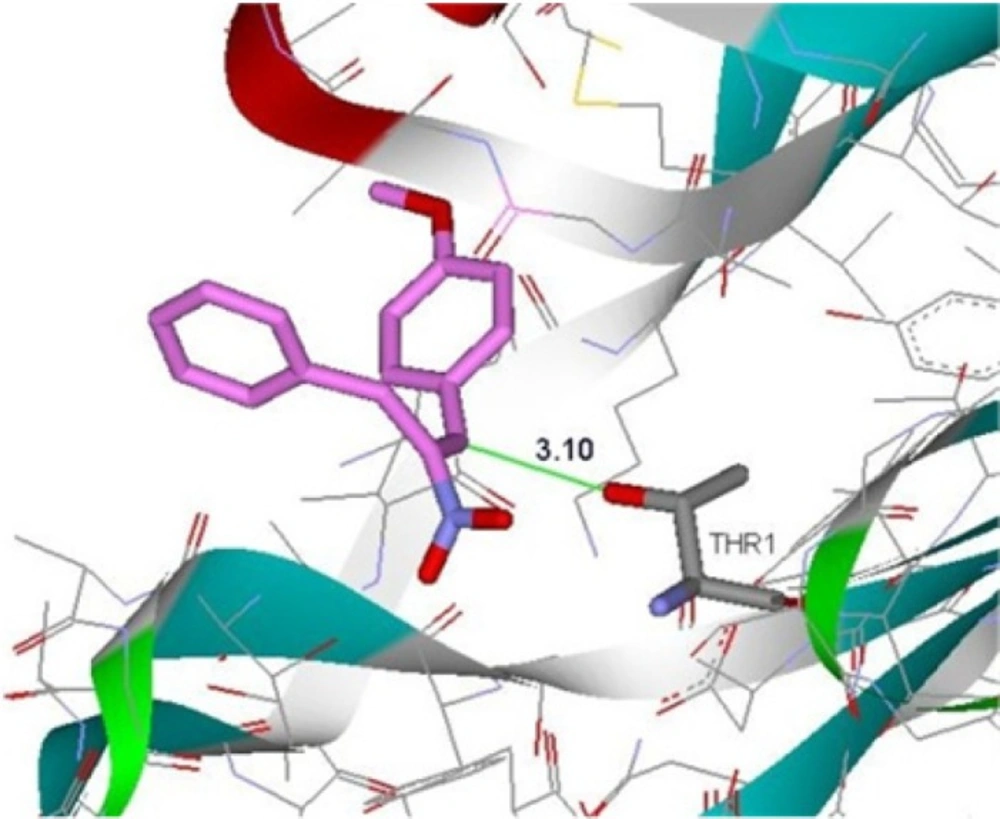
Binding of compound 4d in the active site of β5-subunit of 20S proteasome
1-methoxy-4-(2-nitrovinyl) benzene (1d): Crystalline (needle shape) yellow solid. Yield: 96%. mp 84-86 °C; IR (KBr): 1624, 1509, 1419, 1324, 973. GC-MS (m/z): 179.1 [M]+. 1H-NMR (400 MHz, CDCl3): 3.87 (3H, s, OMe), 6.95 (2H; d, J = 8.8 Hz; CHAr-o-OMe), 7.49-7.54 (3H, m, CHAr-m-OMe and =CHα), 7.97 (1H; d, J = 13.6 Hz; =CHβ). Anal. Calcd. For C9H9NO3 (179.17): C, 60.33; H, 5.06; N, 7.82; O, 26.79. Found: C, 60.31; H, 5.08; N, 7.80; O, 26.79.
N, N-dimethyl-4-(2-nitrovinyl)benzenamine (1e): Red solid. Yield: 30%. mp 182-184 °C; IR (KBr): 1668, 1547, 1493, 1339, 981. GC-MS (m/z): 192.1 [M]+. 1H-NMR (400 MHz, CDCl3): 3.08 (6H, s, CHN(Me)2), 6.9 (2H; d, J = 8.9 Hz; CHAr-o-N(Me)2), 7.43 (2H; d, J = 8.9 Hz; CHAr-m-N(Me)2), 7.50 (1H, d, J = 13.4 Hz; =CHα), 7.97 (1H, d, J = 13.4 Hz; =CHβ). Anal. Calcd. For C10H12N2O2 (192.21): C, 62.49; H, 6.29; N, 14.57; O, 16.65. Found: C, 62.48; H, 6.29; N, 14.55; O, 16.66.
General procedure for the synthesis of 2-nitro-3-arylprop-2-en-1-ol (2a-d)
To a stirred solution of substituted nitrostyrene (10 mmol) in THF (25 mL) at room temperature, imidazole (1 equivalent) and anthranilic acid (10 mol%) were added. Aqueous solution of formaldehyde (37%, 25 mL) was then added and the mixture was stirred at room temperature overnight. After completion of the reaction (monitored by TLC), the mixture was acidified with 5 M HCl (20 mL) and the aqueous layer was extracted with EtOAc (3 × 25 mL). The organic layers were washed with brine (50 mL), dried with anhydrous Na
2SO
4, and concentrated .The residue was purified by column chromatography (silica gel, EtOAc–hexanes, 0–30%, gradient elution) to obtain pure product (
16).
2-Nitro-3-phenylprop-2-en-1-ol (2a): Yellow oil. Yield: 53%. IR (neat): 3437, 1658, 1531, 1238, 1027, 783. GC-MS (m/z): 179.1 [M]+. 1H-NMR (400 MHz, CDCl3): 2.67 (1H, s, br), 4.72 (2H, s, CH2), 7.48-7.58 (5H, m, CHPh), 8.22 (1H, s, =CH). Anal. Calcd. For C9H9NO3 (179.17): C, 60.33; H, 5.06; N, 7.82; O, 26.79. Found: C, 60.35; H, 5.06; N, 7.79; O, 26.80.
2-Nitro-3-(4-chlorophenyl)prop-2-en-1-ol (2b): Pale yellow oil. Yield: 32%. IR (neat): 3441, 1636, 1506, 1453, 1091, 841. GC-MS (m/z): 213.0 [M]+. 1H-NMR (400 MHz, CDCl3): 2.66 (1H, s, br), 4.67 (2H, s, CH2), 7.46 (2H; d, J = 8 Hz; CHAr-o-Cl), 7.52 (2H; d, J = 8 Hz; CHAr-m-Cl), 8.16 (1H, s, =CH). Anal. Calcd. For C9H8ClNO3 (213.62): C, 50.60; H, 3.77; Cl, 16.60; N, 6.56; O, 22.47. Found: C, 50.59; H, 3.78; Cl, 16.62; N, 6.54; O, 22.46.
2-Nitro-3-(4-tolyl)prop-2-en-1-ol (2c): Yellow solid. Yield: 38%. mp 52–54 °C. IR (KBr): 3444, 1660, 1539, 1342, 1030, 833. GC-MS (m/z): 193.1 [M]+. 1H-NMR (400 MHz, CDCl3): 2.42 (3H, s, Me), 2.67 (1H, s, br), 4.71 (2H; s, CH2), 7.29 (2H; d, J = 8 Hz; CHAr-o-Me), 7.47 (2H; d, J = 8 Hz; CHAr-m-Me), 8.19 (1H, s, =CH). Anal. Calcd. For C10H11NO3 (193.2): C, 62.17; H, 5.74; N, 7.25; O, 24.84. Found: C, 62.15; H, 5.74; N, 7.23; O, 24.85.
2-Nitro-3-(4-methoxyphenyl)prop-2-en-1-ol (2d): Yellow solid. Yield: 54%. mp 71–73 °C. IR (KBr): 3424, 1636, 1512, 1382, 1024, 831. GC-MS (m/z): 209.1 [M]+. 1H-NMR (400 MHz, CDCl3): 2.62 (1H, s, br), 3.48 (3H, s, OMe), 4.73 (2H, s, CH2), 6.99 (2H; d, J = 8 Hz; CHAr-o-OMe), 7.50 (2H; d, J = 8 Hz; CHAr-m-OMe), 8.19 (1H, s, =CH). Anal. Calcd. For C10H11NO4 (209.2): C, 57.41; H, 5.30; N, 6.70; O, 30.59. Found: C, 57.43; H, 5.31; N, 6.69; O, 30.60.
Synthesis of (2-nitroethyl)benzene (3)
To the stirred mixture of nitrostyrene (1a, 10 mmol), silica gel (20 g, column chromatography grade), 2-propanol (30 mL), and chloroform (160 mL) was added NaBH
4 (40 mmol) in small portions over a period of 15 min at room temperature. The mixture was stirred for additional 15 min until the yellow color was completely disappeared. Excess NaBH
4 was decomposed by addition of diluted HCl (0.1 N) and the mixture was filtered and washed with CH
2C1
2. The combined filtrate was washed with brine, dried (Na
2SO
4) and then evaporated to give (2-nitroethyl)benzene as a clear oil (
17).
IR (neat): 3063, 3031, 1608, 1555, 1380, 752. 1H-NMR (400 MHz, CDCl3): 5.44 (2H, s, CH2), 7.45-7.50 (5H, m, CHPh). Anal. Calcd. For C8H9NO2 (151.16): C, 63.56; H, 6.00; N, 9.27; O, 21.17. Found: C, 63.55; H, 6.02; N, 9.26; O, 21.14.
General procedure for the synthesis of aryl-2-nitroallylbenzene (4a-4e)
A mixture of (2-nitroethyl)benzene (15 mmol), dimethylamine hydrochloride (30 mmol), potassium fluoride (2.25 mmol) and appropriate aldehyde (15 mmol) in toluene (20 mL) was refluxed with a Dean–Stark trap for 24 h. The reaction mixture was then diluted with toluene (100 mL) and washed with 10% HCl solution (3 × 30 mL). The organic phase was dried over anhydrous Na
2SO
4 and solvent evaporated
in-vacuo. The product was purified by recrystallization in a mixture of EtOAc/hexane or column chromatography over silica gel (EtOAc/hexane, 7/3) (
18).
3-(phenyl)-2-nitroallylbenzene (4a): Yellow oil. Yield: 67%. IR (neat): 1649, 1533, 1392, 1331, 779. GC-MS (m/z): 239.1 [M]+. 1H-NMR (400 MHz, CDCl3): 4.27 (2H, s, CH2), 7.21-7.27 (5H, m, CHbenzyl ring), 7.42-7.47 (5H, m, CHPh), 8.31 (1H, s, =CH). Anal. Calcd. For C15H13NO2 (239.27): C, 75.30; H, 5.48; N, 5.85; O, 13.37. Found: C, 75.28; H, 5.49; N, 5.83; O, 13.35.
3-(4-chlorophenyl)-2-nitroallylbenzene (4b): Light brown solid. Yield: 78%. mp 98–100 °C. IR (KBr): 1651, 1520, 1325, 1021, 877. GC-MS (m/z): 273.1 [M]+. 1H-NMR (400 MHz, CDCl3): 4.22 (2H, s, CH2), 7.14 (2H; d, J = 8.4 Hz; CHAr-o-Cl), 7.29 (2H; d, J = 8.4 Hz; CHAr-m-Cl), 7.42-7.44 (5H, m, CHPh), 8.31 (1H, s, =CH). Anal. Calcd. For C15H12ClNO2 (273.71): C, 65.82; H, 4.42; Cl, 12.95; N, 5.12; O, 11.69. Found: C, 65.80; H, 4.40; Cl, 12.94; N, 5.14; O, 11.70.
3-(4-methylphenyl)-2-nitroallylbenzene (4c): Yellow-green solid. Yield: 73%. mp 105–107 °C. IR (KBr): 1665, 1520, 1393, 1329, 782. GC-MS (m/z): 253.1 [M]+. 1H-NMR (400 MHz, CDCl3): 2.32 (3H, s, Me), 4.22 (2H, s, CH2), 7.09-7.14 (4H, m, CHAr-o-Me and CHAr-m-Me), 7.40-7.46 (5H, m, CHPh), 8.28 (1H, s, =CH). Anal. Calcd. For C16H15NO2 (253.3): C, 75.87; H, 5.97; N, 5.53; O, 12.63. Found: C, 75.88; H, 5.96; N, 5.53; O, 12.61.
3-(4-methoxyphenyl)-2-nitroallylbenzene (4d): Yellow solid. Yield: 57%. mp 53–56 °C. IR (KBr): 1661, 1523, 1342, 1303, 878. GC-MS (m/z): 269.1 [M]+. 1H-NMR (400 MHz, CDCl3): 3.85 (3H, s, OMe), 4.31 (2H, s, CH2), 6.95 (2H; d, J = 8 Hz; CHAr-o-OMe), 7.20-7.39 (5H, m, CHPh), 7.45 (2H; d, J = 8 Hz, CHAr-m-OMe), 8.35 (1H, s, =CH). Anal. Calcd. For C16H15NO3 (269.3): C, 71.36; H, 5.61; N, 5.20; O, 17.82. Found: C, 71.35; H, 5.62; N, 5.23; O, 17.80.
N, N-dimethyl-4-(2-nitro-3-phenylprop-1-enyl)benzenamine (4e): Dark red solid. Yield: 76%. mp 110–113 °C. IR (KBr): 1599, 1530, 1386, 1252, 875. GC-MS (m/z): 282.1 [M]+. 1H-NMR (400 MHz, CDCl3): 3.02 (6H, s, N(Me)2), 4.34 (2H, s, CH2), 6.65 (2H; d, J = 9 Hz; CHAr-o-N(Me)2), 7.22-7.34 (5H, m, CHPh), 7.40 (2H; d, J = 9 Hz, CHAr-m- N(Me)2), 8.35 (1H, s, =CH). Anal. Calcd. For C17H18N2O2 (282.34): C, 72.32; H, 6.43; N, 9.92; O, 11.33. Found: C, 72.30; H, 6.44; N, 9.94; O, 11.32.
Synthesis of 3-methyl-1-nitrobutane (5)
Isopentyl iodide (50.0 mmol) dissolved in DMF (40 mL) was added dropwise to a stirred solution of DMF (150 mL) and sodium nitrite (100.0 mmol) at 0 °C. The mixture was stirred overnight at room temperature. Water (500 mL) was then added to the mixture and it was extracted with petroleum ether (4 × 70 mL). The combined organic phase was washed with water (2 × 50 mL) and dried with Na2SO4. The solvent was evaporated under reduced pressure to give 3-methyl-1-nitrobutane as colorless oil.
IR (neat): 2962, 2870, 1785, 1555, 1473, 1275, 1135. 1H-NMR (400 MHz, CDCl3): 0.89 (6H; d, J = 6.6 Hz; -CH3-isopropyl), 1.58-1.66 (1H, m, -CH-isopropyl), 1.85 (2H; q, J = 7.3 Hz; -CHβ), 4.34 (2H; t, J = 7.3 Hz, CH2). Anal. Calcd. For C5H11NO2 (117.15): C, 51.26; H, 9.46; N, 11.96; O, 27.32. Found: C, 51.27; H, 9.46; N, 11.95; O, 27.30.
General procedure for the synthesis of Aryl-4-methyl-2-nitropent-1-ene (6a-b)
To a stirred solution of 3-methyl-1-nitrobutane (28 mmol) and appropriate aldehyde (28 mmol) in MeOH (20 mL), AcOH (1.08 mL, 18.8 mmol) and n-BuNH
2 (1.86 mL, 18.8 mmol) were added at 0 °C under argon. After being stirred for 36 h at room temperature, the reaction mixture was poured into water and then extracted with EtOAc. The organic layer was washed with water and brine, dried over Na
2SO
4 and concentrated. The resultant residue was purified by column chromatography on silica gel (EtOAc/hexane, 1/20) (
19).
1-methyl-4-(4-methyl-2-nitropent-1-enyl)benzene (6a): Colorless oil. Yield: 40%. IR (neat): 1640, 1524, 1384, 1188, 770. GC-MS (m/z): 219.1 [M]+. 1H-NMR (400 MHz, CDCl3): 0.96 (6H; d, J = 6.8 Hz; -(CH3)2-isopropyl), 2.1 (1H; n, J = 6.8 Hz; -CH-isopropyl), 2.40 (3H, s, Me), 2.83 (2H; d, J = 7.2 Hz; CH2), 7.25 (2H; d, J = 8 Hz; CHAr-o-Me), 7.36 (2H; d, J = 8 Hz; CHAr-m-Me), 8.04 (1H, s, =CH). Anal. Calcd. For C13H17NO2 (219.28): C, 71.21; H, 7.81; N, 6.39; O, 14.59. Found: C, 71.19; H, 7.80; N, 6.39; O, 14.60.
1-methoxy-4-(4-methyl-2-nitropent-1-enyl)benzene (6b): Yellow oil. Yield: 44%. IR (neat): 1599, 1506, 1302, 1025, 832. GC-MS (m/z): 235.1 [M]+. 1H-NMR (400 MHz, CDCl3): 0.95 (6H; d, J = 6.8 Hz; -(CH3)2-isopropyl), 2.0 (1H; n, J = 6.8 Hz; -CH-isopropyl), 2.42 (2H; d, J = 7.2 Hz; CH2), 3.86 (3H, s, OMe), 6.97 (2H; d, J = 8 Hz; CHAr-o-OMe), 7.42 (2H; d, J = 8 Hz; CHAr-m-OMe), 8.05 (1H, s, =CH). Anal. Calcd. For C13H17NO3 (235.28): C, 66.36; H, 7.28; N, 5.95; O, 20.40. Found: C, 66.35; H, 7.29; N, 5.96; O, 20.39.
Cell viability assays
Human breast cancer (MCF-7), and human prostate cancer (PC-3) cells were cultured in their respective media supplemented with 10% fetal bovine serum (FBS). The cells were cultured at 1 × 104 cells per well, in a 96-well plate and incubated for 24-48 h to obtain cell attachments. Afterwards, the cells were treated with different concentrations of test compounds for 48 h and then incubated with 20 µL of 5 mg/mL 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) solution for 4 h. After aspiration of the MTT solution, 100 µL of DMSO was added to each well to dissolve formazan crystals. The absorbance of each well was measured at the wavelength of 550 nm using an enzyme-linked immunosorbent assay reader. IC50 values were calculated from the curves generated by plotting the percentage of the viable cells versus the test concentration on a logarithmic scale using SigmaPlot 10.0 software.
Cell-based proteasome inhibition assay
The effect of the derivatives on the 20S proteasome activity was determined using the 20S proteasome activity assay kit from Sigma (USA). This assay measures the ChT-L protease activity in cultured cells and is based on the detection of the fluorophore R110 after cleavage from the labeled substrate LLVY-R110. MCF-7 and PC-3 cells were pretreated with different concentrations of corresponding compounds and proteasome activities were determined according to the manufacturer′s protocol. Proteasome activity generates strongly green fluorescent R110, which was monitored at an excitation wavelength of 480 nm and emission of 590 nm on a microplate fluorescence reader. All data were calculated by subtracting the blank control. The data are presented as percent inhibition at a specific concentration (10 µM for MCF-7 and 25 µM for PC-3), and IC50 values obtained from dose–response curves utilizing curve-fitting software package SigmaPlot 10.0 software.
Docking studies
The crystal structure of 20S proteasome (PDB code: 4INR) was obtained from the RCSB Protein Data Bank. The molecular docking study was performed using AutoDock Vina v.1.1.2. Protein was prepared for docking by removing co-crystallized ligand and all water molecules from crystal protein. Polar hydrogens were added and nonpolar hydrogens were merged, finally Kallman unitedatom charge and atom type parameter were added to 4INR. Grid box dimensions (20 × 20 × 20) were set surrounding active site (
20).