To our knowledge, this is the first study to compare the combination of bolus and continuous infusion of hypertonic saline plus furosemide versus hypertonic saline alone in patient with TBI, and to compare sNGAL and sCr as marker for diagnostic and monitoring purposes. This study’s most notable finding was that sNGAL and sCr concentrations decreased in the HTS+F group, and also blood pH level decreased significantly in the HTS group. Other important finding is that the incidence of AKI (KDIGO criteria) in the HTS group was 4-fold higher than in the HTS+F group; although there was not a statistically significant difference, incidence of AKI according to sNGAL concentrations >150 ng/mL in the HTS group is statistically higher than in the HTS+F group.
Over the past decade sNGAL has become a marker for early detection of AKI, as its concentration increases within a few hours post-insult. Serum or urine levels of NGAL correlate with the severity of AKI. Patients with elevated sNGAL concentrations, even those with normal sCr concentrations, seem to have an increased risk of mortality or of requiring renal replacement therapy (
3-
5). McIlroy
et al. suggested that chronic kidney injury reduced the accuracy of sNGAL in predicting AKI, with sNGAL being best identifying AKI in patients with a baseline glomerular filtration rate (GFR) of 90–120 mL/min (
17). As our patients had normal renal function (GFR: 80–120 mL/min) at baseline, they were almost homogeneous and good choice for evaluating sNGAL. There is no consensus agreement regarding NGAL cut-off point to define AKI, but several sNGAL cut-off points are used to determine AKI. Some studies suggest that sNGAL concentrations <100 ng/mL and >150 ng/mL are useful to exclude and diagnose AKI, respectively (
3,
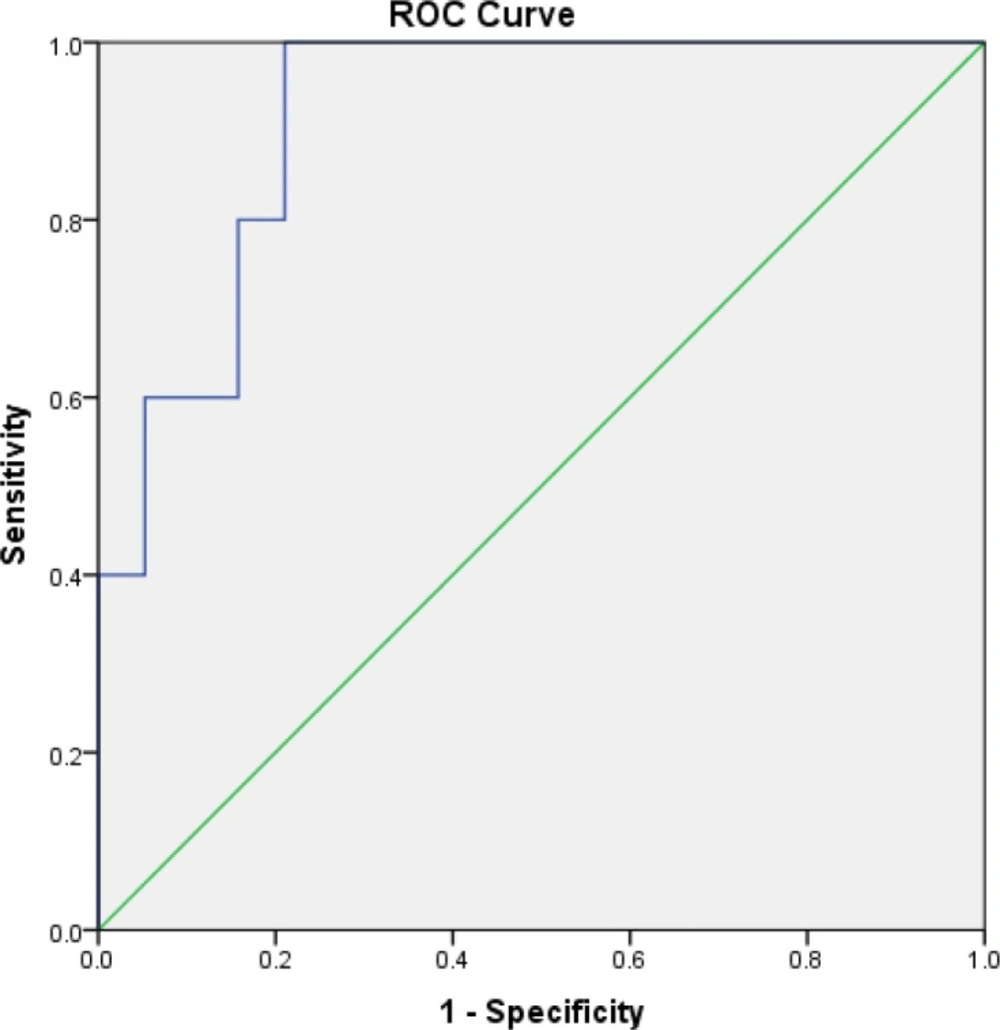
16). We chose a cut-off of >150 ng/mL as a potential risk factor for kidney injury. In our study, all patients who developed stage one AKI (KDIGO criteria) had baseline sNGAL concentrations of >150 ng/mL, and sNGAL increased earlier than sCr did.
Although the sNGAL concentration displayed greater variation than did the sCr concentration at baseline, it indicates that TBI can lead to renal damage or systemic generation of sNGAL as a stress reaction, regardless of the type of treatment administered. This finding is in keeping with those of previous studies (
1,
5). It can explained by brain–kidney cross-talk (
18) (brain damage progresses to the syndrome of multiple organ dysfunction, a syndrome that is likely mediated by dysregulated inflammatory mechanisms) and rise of sNGAL after inflammatory condition (
4,
19).
Although the level of sNGAL was >150 ng/mL in some patients at baseline, after our intervention, the sNGAL concentration significantly decreased in the HTS+F group but increased in the HTS group. It seems that this was related not only to TBI but also to other factors.
Changes in sCr concentrations are more reliable than sNGAL, because there is no consensus agreement for NGAL cut-off point, sCr is not affected by environment stress and systemic generation. In our study, sCr concentrations increased in the HTS group and it was significantly different on day 2 and 3 compared to HTS+F group.
The use of mannitol and hypertonic saline, coagulopathy, sepsis, rhabdomyolysis, blood loss, contrast agents and antibiotics was associated with rise sCr and risk of kidney injury
Many studies have shown hypertonic saline to be therapeutically beneficial in TBI, but its effects on the kidney remain controversial (
6,
8,
9 and
23). Hypertonic saline (especially continuous infusion), and chloride-containing solutions can cause systemic complications such as hyperchloremia, metabolic acidosis, and AKI (
9,
18). Kelly
et al. investigated the effect of hypertonic saline on patients with severe TBI (
19). AKI occurred in 12.1% of patients in the continuous infusion group but did not occur in the bolus group. In another study, patients with burns were treated with hypertonic saline or Ringer lactate solutions; renal failure and mortality were 4 and 2 times higher, respectively, in the group treated with hypertonic saline (
24).
The mechanisms by which hypertonic saline induces kidney injury are unclear, but several hypotheses have been proposed. First, chloride-containing solutions, via mesangial contraction, increase chlorine absorption and induce renal vasoconstriction (
18,
24 and
25). Chloride also induces thromboxane production; thromboxane causes renal vasoconstriction and reduced renal perfusion delays the onset of diuresis (
9). Second, it may increase the risk of hypernatremia and hyperchloremia, causing an osmotic gradient resulting in fluid shifts from the endothelial cell to the interstitial space (
6,
24 and
26). This hypothesis is acceptable as low chloride solutions reduce the AKI risk in ICU patients regardless of the underlying disease (
9).
To date, none have evaluated this combination in humans as a form clinical trial. A few studies have combined furosemide with hypertonic saline or mannitol in rats and metabolic acidosis was reported in the hypertonic saline group versus furosemide with hypertonic saline (
27). In our study, blood pH decreased in both groups, but was significantly lower in the HTS than in the HTS+F group. This hypothesis can be proven that hypertonic saline-induced acidosis may lead to an increase in sNGAL and sCr concentrations. Patients with high baseline levels of sNGAL following TBI were more sensitive to develop hypertonic saline-induced metabolic acidosis, but concomitant administration of furosemide and hypertonic saline had positive effects on the kidney and reduced the concentration of sCr and sNGAL; however, no significantly difference in outcome was found between the 2 groups.
Several studies have demonstrated the effects of furosemide on the kidney (
10,
12). One meta-analysis found that the combination of furosemide and hydration reduced contrast-induced AKI in patients undergoing angiography (
12); another found that furosemide may be more effective in mild AKI than in severe AKI (
10). In terms of evidence-based pharmacology, loop diuretics act on the Na
+/K
+/2Cl
− cotransporter in loop of Henle, thereby reducing oxygen demand in medulla. Furosemide also improves renal cytochrome oxygenation, and enhances the rate of elimination of toxins and casts from the tubules. Furosemide stimulates prostaglandin production and reduces ischemia via renal vasodilation. Finally, in contrast to hypertonic saline-induced hyperchloremic metabolic acidosis, furosemide causes a metabolic alkalosis (
10,
11,
28 and
29). Therefore, furosemide may reduce hypertonic saline induced kidney injury via inducing a metabolic alkalosis and via other mechanisms mentioned.
We also identified clindamycin as a risk factor; few studies have reported an association between clindamycin and AKI (
30). Hypokalemia was the most common adverse event in our study
. Disruption of sodium and potassium exchange in the distal tubule, caused by both hypertonic saline and furosemide, leads to hypokalemia (
28,
31).
Our study has some limitations. First, it was restricted to one center, the data were open-label, and the clinicians and data collectors were not blinded. Second, sNGAL is also a stress marker; its concentration increases after infectious, inflammatory, and ischemic insults. Third, some researchers have shown that urinary NGAL may be a better marker of intrinsic AKI than sNGAL, but we were not able to measure urinary NGAL concentrations. Fourth, as mentioned before, there is no consensus agreement regarding NGAL cut-off point to define AKI. Changes in its serum concentrations (especially urine concentrations) are more reliable. A cut-off value of 150 ng/mL for sNGAL concentration seems arbitrary albeit reasonable, but our findings may have been different had we chosen a different cut-off value. Fifth, the incidence of AKI may have been underestimated; because of we excluded patients with renal impairment to improve the reliability of sNGAL in evaluating AKI. Lastly, the study does not reflect the effect of hypertonic saline plus furosemide on preventing AKI—this needs further exploration and larger sample size.
In conclusion, TBI can lead to an early increase in sNGAL concentration. The use of hypertonic saline plus furosemide, compared with hypertonic saline alone, was associated with lower sNGAL and sCr concentrations. There was no significant difference between groups regarding AKI incidence and using hypertonic saline plus furosemide in preventing AKI requires further research.