Instrumentation
Spectrophotometric measurements were performed by using Shimadzu RF–1501 UV-VIS (ultraviolet-visible spectrophotometer). Anticancer (cytotoxic) activities of samples were spectrophotometrically measured by well known MTT assay (Microplate spectrophotometer system (BioTek® Epoch Microplate Spectrophotometer, Winooski-USA). Fluorescent Inverted Microscope (Olympus), Olympus CKX41 microscope, refrigerated centrifuge (Hettich Universal 30 RF), CO2 incubator (5% CO2, 95% humidity and 37 ºC) (Labotect) were used for Alkaline Comet Assay method.
Collection of plant material
Plants were sampled from Mardin-Ömerli road 4 Km (Zınnar valley) on flowering, fruiting and seedings stage from April to July. Plants materials were maintained at the Mardin Artuklu University Herbarium (2014-3-4-5-MAU). Plants were identified and authenticated by Dr. Cumali Keskin.
Preparation of plant methanol extracts
The 100 g plant materials were dehydrated in the shade (25 ± 2 °C) for 10 days. A total of 20 g of each material was grounded in a grinder with a 2 mm diameter mesh and incubated with 200 mL (99%) methanol at room temperature for 3 days. The obtained extracts were filtered with Whatman No.1 filter paper and methanol phase was removed on rotary evaporator under vacuum. Nearly, 2 g of the crude methanol extracts were obtained from each part of the plant material. The extracts were kept in dark and airtight glass bottles at -20 ºC until used for experimental studies.
Determination of total phenolic content
Folin-Ciocalteu method applied to methanol extracts to determine their total phenolic contents (
21). Absorbance of tested solutions was measured at 765 nm. Results were expressed as gallic acid equivalents (GAE) µg GAE/g extract of dry plant material) using the following equation obtained from a linear curve that gallic acid used a standard (y = 0.008X+0.004, R
2 = 0.9980).
Chromatographic analyses
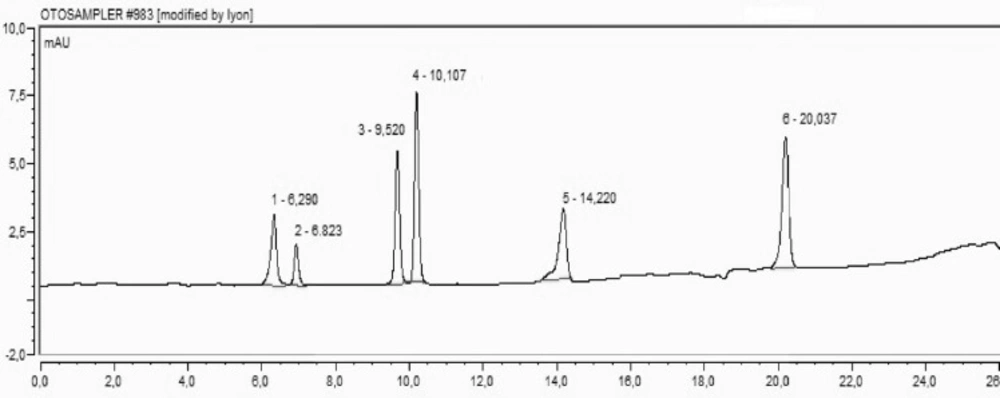
The methanol extracts of different parts of H. retusum were analysed for their phenolic compounds by HPLC (Agilent 1260 Infinity HPLC-DAD with Chem Station revision B.04.01 software). Phenolic compounds were separated with the Agillent ZORBAX reverse phase C18 column (250 x 4,6- 5 μM) thermostated at 35 °C. Methanol and acetic acid–water (2:98 v/v) were used for gradient elution. Flowers, fruits, and seeds crude extracts (10 mg) were dissolved in methanol (10 mL) before injection to HPLC (20 μL). The phenolic compounds were monitored at 280 nm wavelength.
Antioxidant Activity Assays (Reducing power activity)
FRAP (ferric reducing antioxidant power) assay (
22) was used to determine the antioxidant activities of plant methanol extract (10-500 mg in 1 mL of ethanol). The absorbance of prepared solutions was measured at 700 nm wavelength. The BHT (butylatedhydroxytoluen) and the BHA (butylatedhydroxyanisole) were used as standard antioxidants.
DPPH Radical Scavenging Activity
The free radical scavenging activity was quantitatively tested using 1,1-diphenyl-2-picryl-hydrazil (DPPH) based on the well-known procedure described in literature (
20). The absorbance was measured at 517 nm wavelength. Following equation was applied to determine the percentages of inhibitions (Dorman, 2004).
Determination of Acetyl/Butyrylcholinesterase inhibitory activity
The method described by Ellman, (
23) was applied for cholinesterase inhibitory activities.
H. retusum fruit flower and seed crude extracts were dissolved in ethanol to give concentrations of 4000 μg/mL. Aliquots of 150 mL of 100 mM sodium phosphate buffer (pH 8.0), 10 μL of sample solution and 20 μL AChE (or BChE) solution were mixed and incubated for 15 min at 25 ºC. Finally, 10 μL of DTNB (5,5-dithio-bis (2-nitrobenzoic acid) and 10 μL acetylthiocholine iodide (or butyryl thiocholine iodide) were added. The last concentrations of the mixture were 200 μg/mL. Inhibition percentages were calculated by using the following equation while galanthamine as a standard drug:
Cell lines, culture treatments
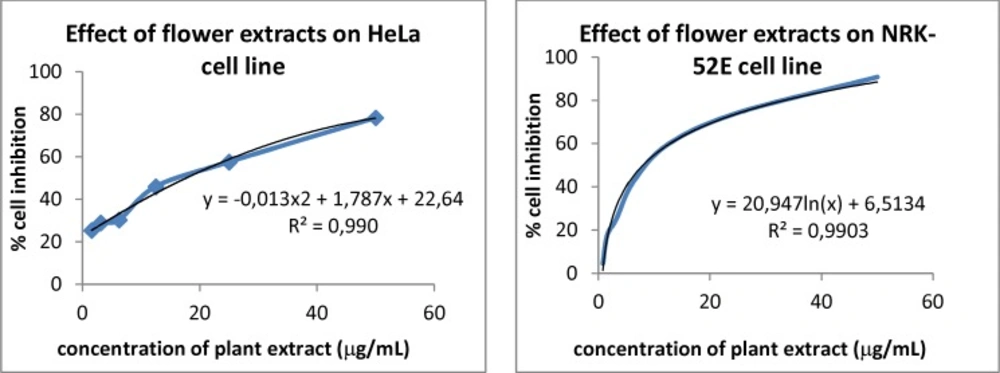
ATCC CCL-2 HeLa (human cervix cancer) and ATCC CRL-1571 NRK-52E lines (rat kidney epithelium cell) cultured by recommended protocols of manufacturers were applied to plants methanol extracts to determine their sensitivities by the MTT colorimetric assay. The cells were seeded at 104 cells/100 µL into each well of 96-well plates and incubated for 24 h at 37 °C and in 5% CO2. Next, the culture medium was abolished and the extracts were added to the wells in different concentrations.
The exposure concentrations were expressed as µg/mL for the plant methanol extracts. Both of cell lines were exposed to plant extracts mentioned above.
MTT cytotoxicity assay
Among the enzyme-based assays, the MTT assay is the best known method to determine the mitochondrial dehydrogenase activities in the living cells. Cytotoxicity assays together with cell viability studies are used for drug screening of chemicals. The method described by Alley
et al. (
24) was applied for cytotoxic activity of plant methanol extracts. In all applied tests the absorbance was measured at 590 nm wavelength. LC
50 values were calculated by using the following equation as the percentages of solvent controls;
Statistical method
All of the samples were subjected to methods in triplicate (n = 3). The results were presented as average ± SD, where SD was standard deviation. Student’s t-test was applied to compare results. A P-value of less than 0.05 was accepted as significant.
Anti-genotoxic Activity
Separation and incubation of mononuclear leukocytes:
20 mL heparinised blood sample was taken from healthy, non-smoking 26-year-old male volunteers. 5 mL Histopaque-1077 were added into four sterile tubes. Successively, 5 mL heparinised blood were added slowly. The tubes were centrifuged at 2100 rpm at 25 degrees for about 30 min. After centrifugation of the lymphocytes, which accumulate in the middle layer of the tube, they were taken into empty tubes with the help of a 1 mL pipette. In order to remove the Histopaque solution 5 mL 1 mol/L phosphate buffer saline (PBS) (pH 7.4) were added to samples before centrifugation at 1600 rpm at 25 degrees for 10 min. The upper layer was removed and the leukocyte pellet was obtained.
Cell viability assay:
Trypan blue stain was used for the evaluation of cell viability assay. The cells were trypsinized and collected from the culture flask. Then, they were mixed with an equal volume (1:1) of trypan blue. They were incubated for 5 min, as well. Stained and unstained cells were counted under the light microscope.
Preparation and incubation of the mononuclear leukocyte suspension
Dulbecco’s modified Eagle’s medium (DMEM) was used for the culture of human mononuclear leukocyte cells. 10% fetal bovine serum (FBS) and 5 μg/mL gentamicin were added to DMEM. Then the prepared cell culture flasks in 5% CO2 atmosphere at 37 °C was dropped into 1, 2 and 3 h of incubation.
The human mononuclear leukocyte cells were used without extract and H2O2 as negative control. 0.7 mM H2O2 treated group without extract administration was used as positive control. The cells were incubated with different concentrations of (10-100 μg/mL) H. retusum methanol extracts.
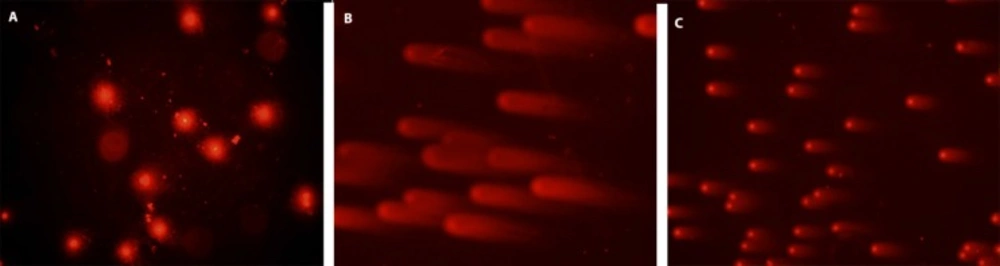
Determination of DNA damage (Alkaline Comet Assay):
The human mononuclear leukocyte DNA damage (hydrogen peroxide induced) was analysed by alkaline comet assay (
25,
26). 80 μL of 0.7% low melting point agarose in PBS was mixed with 10 µL (around 20,000 cells) of cell suspension treated with different concentrations of
H. retusum flower, fruit and seed methanol extract at 37 °C.