Materials
DMEM with 2 mM L-glutamine (Gibco) was supplemented with 3.7 g/L NaHCO3, 10% heat-inactivated fetal calf serum (Faculty of veterinary medicine, University of Tehran) 30 mg/L asparagine, 100 U/mL penicillin and 10 μg/mL streptomycin (Gibco). Anti-PARP (85 kDa fragment) antibody was purchased from Abcam. Trypan blue, MTT, proteinase K, ECoR1-Hind III digested DNA marker, cocktail protease inhibitor, HRP-conjugated IgG, ethidium bromide, cytochrome C, superoxide dismutase were purchased from Sigma-Aldrich chemical company. DPA and other chemical compounds were purchased from Merck. Annexin-V-FLUOS Staining Kit was obtained from Roche. PARP antibody and IgG2a-PE and IgG2b-PE were from AbCam. CD34-PE, C-kit-PE, Sca-1-PE antibodies were a gift from Malek-Ashtar University (Tehran-Iran).
Daunorubicin and doxorubicin were purchased from Helale Ahmar pharmacy (Tehran, Iran, manufactured by Pharmacia) and used without further purification. Stock solutions of the drugs were prepared in sterile distilled water at a concentration of 2 mg/mL and stored at -20 °C in the dark. Before use they were diluted to desired concentrations with PBS, pH 7.4.
Male balb/c mice weighting 20-25 g (6 to 8 weeks old) were obtained from laboratory animal center of IBB. They were maintained in conventional pathogen free conditions in a temperature (22-23 °C), humidity (50-70%), and photoperiod (12 h dark/light cycle) controlled room.
Isolation of multipotent hematopoietic cells of bone marrow and Immunophenotyping
Total bone marrow from each femur and tibia pair was eluted aseptically with 1mL syringe (25 gauge needle) into 1 mL DMEM and aliquot of the cell suspension was diluted in 3% acetic acid to lyse red blood cells. The total nucleated cells were counted and the viability determined by trypan blue exclusion assay. The bone marrow cells were cultured overnight in DMEM at a density of 10
6 cells per mL at 37 °C with 5% CO
2 and fully humidified condition (
18). Non-adherent cells which are the population of hematopoietic stem and progenitor cells were collected and used for further investigation.
For immunophenotyping, 50 μL of PBS (pH 7.4) and the following FITC-conjugated monoclonal antibodies, CD34-PE, C-kit-PE, Sca-1-PE, IgG2a-PE and IgG2b-PE, were added to the cells. After 30 min incubation, the cells were rinsed with 2 mL of PBS and finally resuspended in 1% paraformaldehyde until the time of data acquisition. The data acquisition and analysis were performed by flow cytometry using Becton-Dickinson FACS calibur and WinMDI software.
Cytotoxicity assay
Nonadherent multipotent hematopoietic cells of bone marrow (106 cells/mL) were cultured in the absence and presence of various concentrations of daunorubicin or doxorubicin for 4 h (time course study revealed that 4 h is the best incubation time) at 37 °C with 5% CO2 and fully humidified condition. Daunorubicin or doxorubicin was added directly at the onset of the culture and the cultures without the drugs were used as control.
Trypan blue: Drug treated and the controls were subjected to trypan blue (0.4% w/v) exclusion assay and the viability of the cells determined using hemocytometer.
MTT assay: The method of Mosmann (
19) was used with some modifications. The MTT assay is based on the cleavage of tetrazolium salt to form blue formazan dye in viable cells. The cells (8 × 10
4 cells/well) were seeded into a 96 well plate (Nunclon, Denmark) and incubated with a series of drugs concentrations for 4 h (to control wells, only culture medium was added). Then 10
μL of MTT (5 mg/mL in H
2O) was added to each well and the cells were incubated at 37 °C with 5% CO
2 for 4 h. The medium was aspirated and replaced with 100
μL DMSO per well in order to dissolve the formed violet formazan crystals in the viable cells.
The plates were gently shaken for 5 min at room temperature to allow complete dissolving of the formazan and the absorbance read at 570 nm using a BioTek microplate reader (USA). The percent of cytotoxicity was calculated: % cytotoxicity (cell death) = (1 – [absorbance of experimental wells/absorbance of the control wells]) × 100.
Fluorescent dye staining
Hoechst 33258 staining assay was performed, based on the nuclear morphology (
20). Multipotent hematopoietic cells were cultured for 4 h in the absence and presence of various concentrations of the drugs as described above. After incubation time, the cells were packed by centrifugation at 1000 × g, washed with PBS and then stained by incubating in 25 μL PBS containing 1 µL of 1 mg/mL stock of Hoechst 33258 at 37 °C for 5 min in the dark. Then the cells were visualized under Zeiss fluorescence microscope with a 40X objective equipped with a digital camera. Apoptotic cells were scored on the basis of nuclear morphology changes, such as chromatin condensation and fragmentation from random fields of view.
Gel electrophoresis and immunoblotting of PARP
After incubation of the cells with various concentrations of daunorubicin or doxorubicin, PARP extraction was performed (
21) by resuspending the cells in sample buffer [containing 62.5 mM Tris (pH 6.8), 4 M urea, 10% glycerol, 2% SDS, 5% b-mercaptoethanol and 0.03% bromophenol blue] and the samples were loaded onto a 12% SDS-polyacrylamide gel as described by Laemmle (
22). The gel was run for 1.5 h at 100 V, stained with 0.1 % coomassie brilliant blue R 250, destained in 10% methanol/acetic acid and photographed.
Western blot: The protein samples were run on 12% polyacrylamide gel at 100 V for protein separation. The proteins were then transferred onto a nitrocellulose membrane (
23). The membrane with the immobilized protein bands was incubated for 1 h at 37 °C with 1% (w/v) gelatin in Tris-NaCl buffer (50 mM Tris-HCl, pH 7.4, 150 mM NaCl), referred to as blocking buffer, and washed three times of 5 min with Tris-NaCl buffer. The membrane was then incubated overnight with overall anti rabbit 85 kDa fragment of PARP at 4 °C. After three times washing the membrane with Tris-NaCl/Tween 20 (0.05%), it was incubated with peroxidase-conjugated goat anti rabbit IgG for 2 h at room temperature. After washing (three times), the membrane was incubated with the substrate solution (1.2 mL of 0.3% 4-chloro-1-naphtol in methanol was mixed with 20 mL Tris-NaCl buffer and 20 mL H
2O
2) for 30 min at 37 °C and the reaction stopped by adding distilled water. The membrane was then dried and photographed.
Quantification of DNA fragmentation
The multipotent hematopoietic cells treated with the drugs were collected by centrifugation at 2000 × g for 10 min at 4 °C. The cells were lysed in 0.7 mL of ice cold lysis buffer (5 mM Tris-HCl pH 8, 20 mM EDTA, and 0.5% triton X-100) by incubation at 37 °C for 3 h and then centrifuged at 10000 × g for 20 min at 4 °C. The pellets were resuspended in 0.5 mL of TE buffer (10 mM Tris-HCl, pH 8, 1 mM EDTA). To the pellets (P) and the supernatants (S), 0.5 mL of 10% trichloroacetic acid (TCA) was added and incubated at room temperature for 10 min. The samples were centrifuged for 20 min at 10000 × g and the pellets were resuspended in 0.5 mL of 5% TCA, followed by incubation at 100 °C for 20 min. Subsequently, to each sample 160 μL of diphenylamine solution (150 mg DPA in 10 mL glacial acetic acid, 150 µL of sulfuric acid and 50 µL acetaldehyde16 µg/mL) was added and incubated overnight at room temperature (
25). Absorbance was measured at 600 nm and the percentage of DNA fragmentation calculated: OD
600 of the supernatant/ [OD
600 of the supernatant + OD
600 of the pellet] × 100.
Superoxide anion release assay
The procedure of Mayo and Curnutte was used (
24). After incubation of multipotent hematopoietic cells in the absence and presence of various concentrations of daunorubicin or doxorubicin, the cells were centrifuged for 10 min at 1000 × g.
The packed cells were washed twice with PBS and then to each sample 200 µL cytochrome C (160 µmol/L) and 200 µL phorbol 12-myristate 13-acetate (10−6 mol/L) were added. Also to the control 17 µL (1 µg/mL) SOD (60 U) was added and the samples were incubated at 37 °C for 15 min and then centrifuged at 6000 × g for 10 min at 4 °C. Superoxide anion production was determined from absorbance reading against the control at 550 nm using UV 260 Shimadzu spectrophotometer.
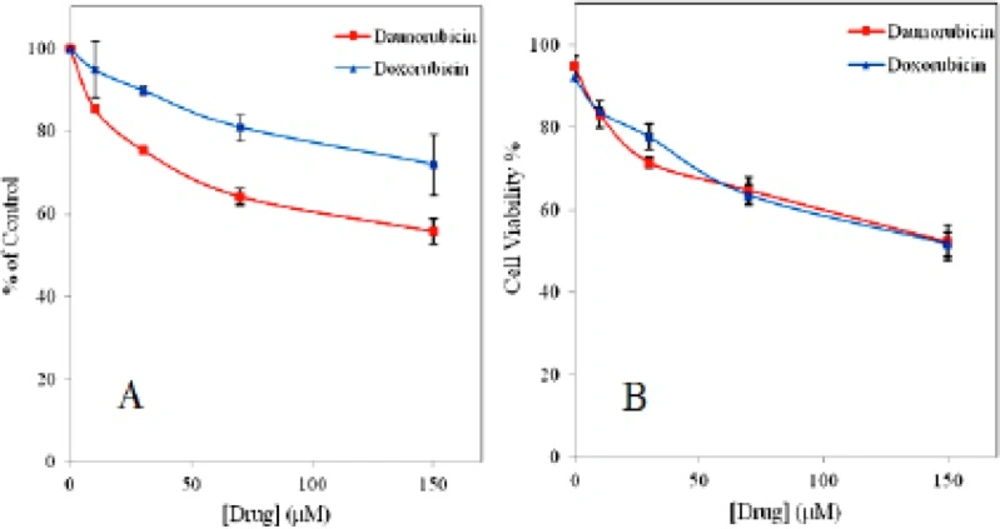
Dose-dependent cytotoxicity of non-adherent multipotent hematopoietic cells treated with various concentrations of daunorubicin and doxorubicin. Cell viability was assessed by trypan blue exclusion (A) and MTT (B) assay. Data are expressed as means ± SD of three independent experiments
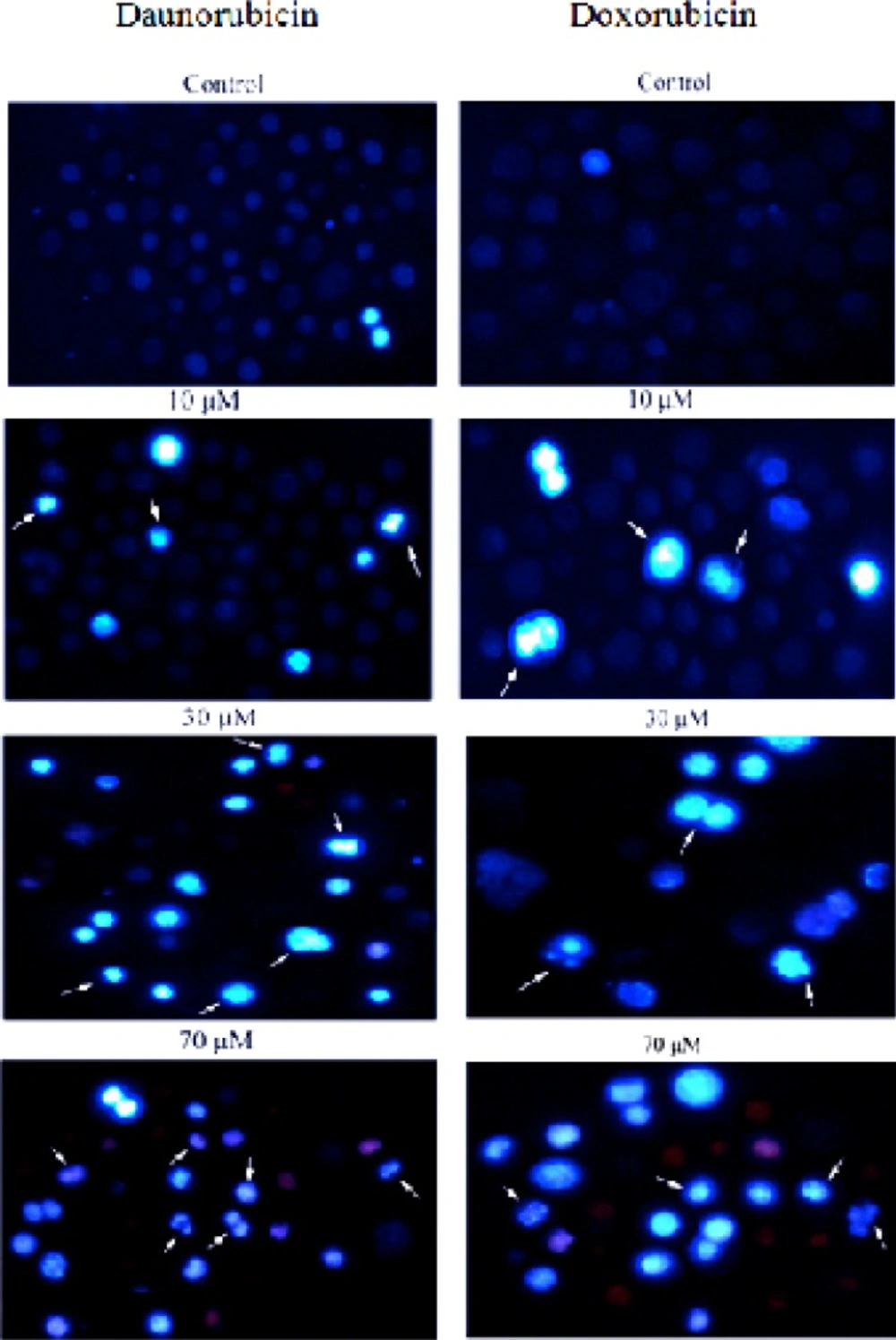
Morphological changes in the non-adherent multipotent hematopoietic cells of mouse bone marrow cells induced in the presence of various concentrations of daunorubicin and Doxorubicin for 4 h and staining with Hoechst 33258. Arrows indicate the cells with condensed chromatin and/or fragmented nuclei. For clarity, only some examples are labeled. (Magnification 200X
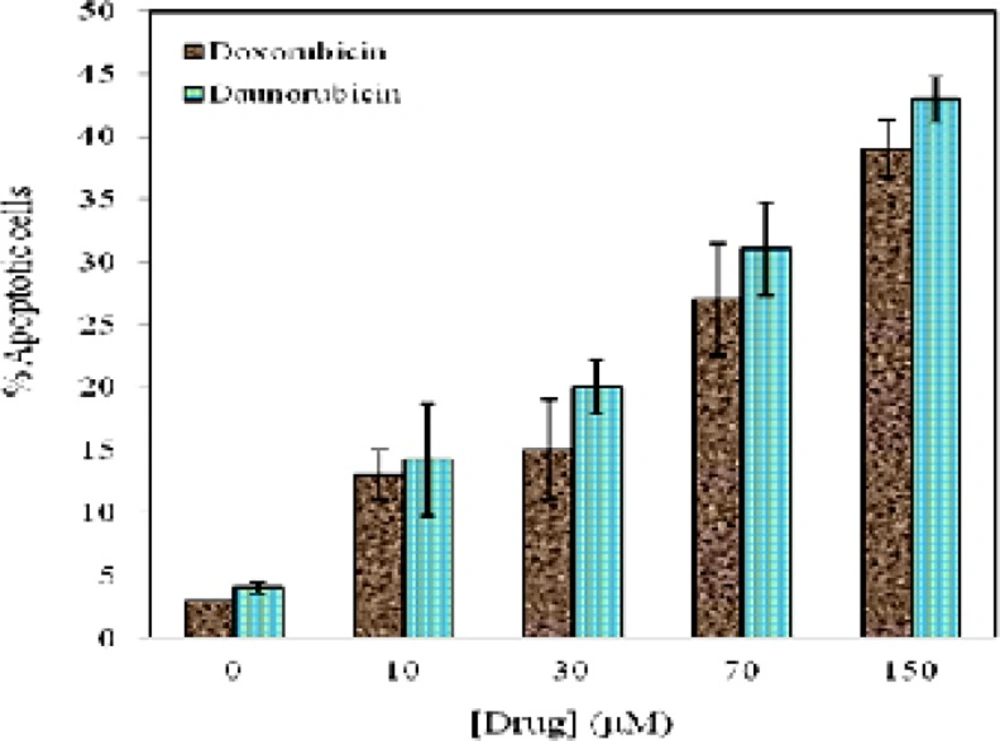
Relative percentage of apoptosis scored on the basis of nuclear morphology changes, such as chromatin condensation and DNA fragmentation from random fields of view for various concentrations of daunorubicin (■) and doxorubicin (■). Data are means ± SD of 3 experiments
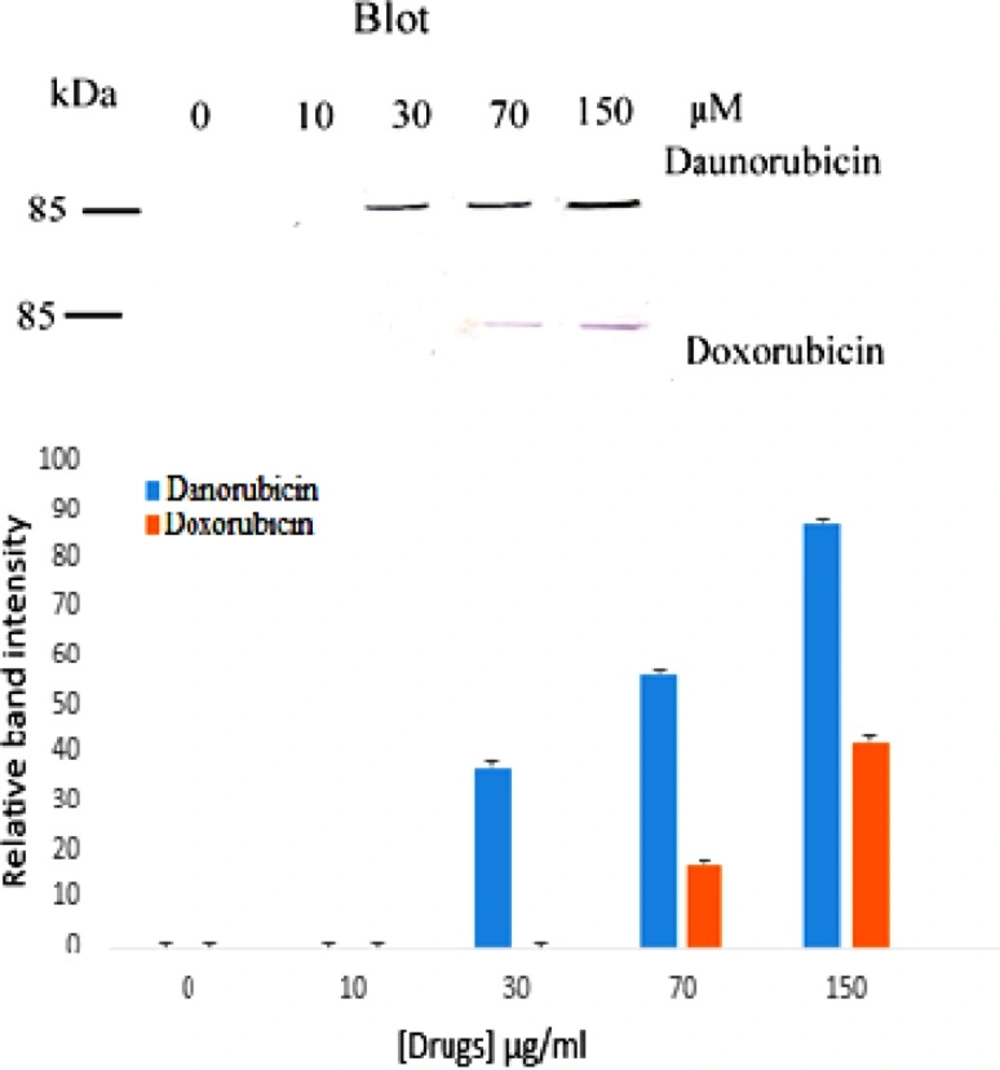
Western blot of PARP cleavage in multipotent hematopoietic cells of bone marrow cells incubated in the absence (Lane 0) and presence of various concentrations of daunorubicin (top) and doxorubicin (bottom) for 4 h. Also the relative band intensity was estimated by Image J is shown. (n = 3).
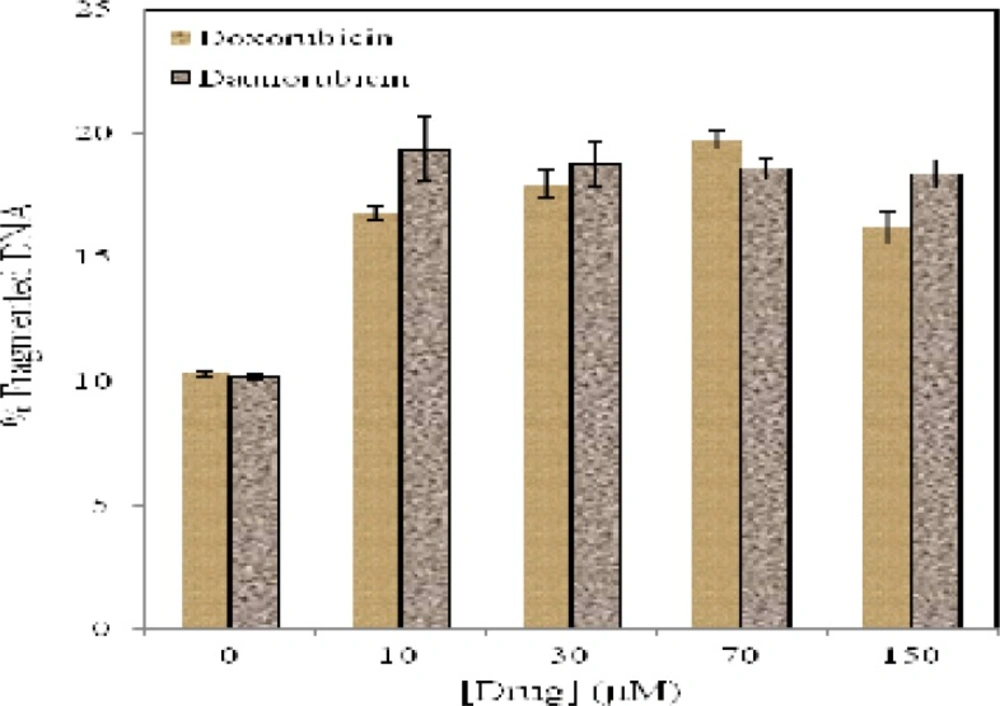
Quantitative estimation of DNA fragmentation in multipotent hematopoietic cells of bone marrow incubated for 4 h in the presence and absence of daunorubicin (■) and doxorubicin (■). Data are means ± SD of 3 experiments (P < 0.05
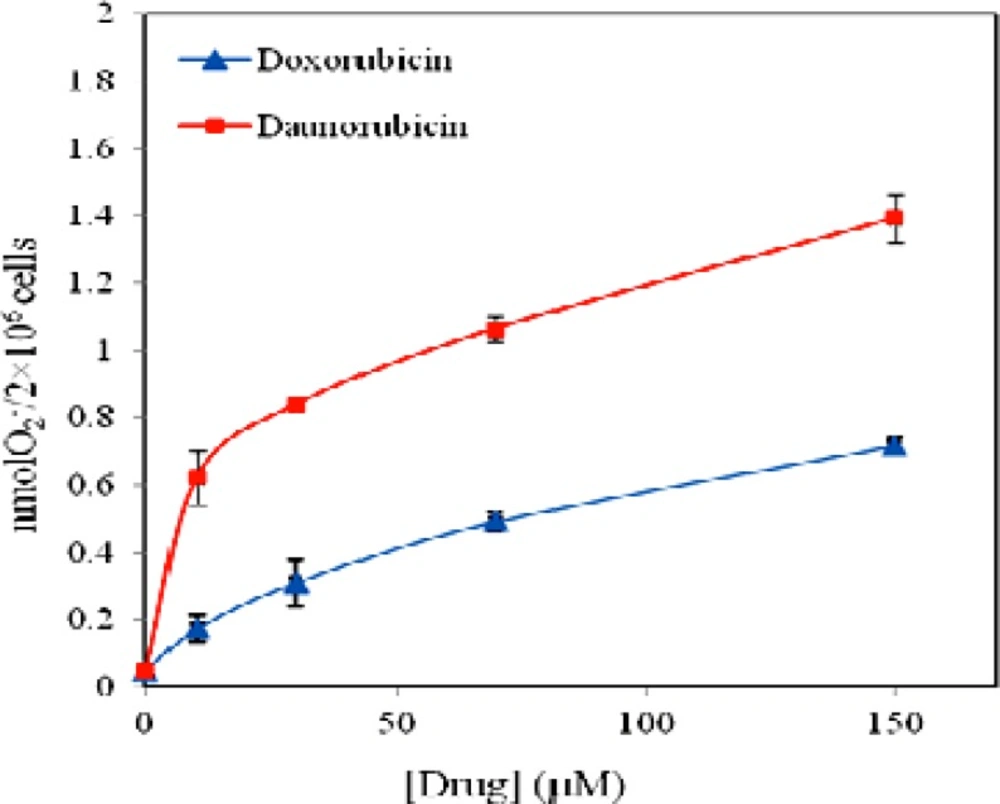
Effect of daunorubicin (■) and doxorubicin (▲) on superoxide anion release from multipotent hematopoietic cells of mouse bone marrow incubated for 4 h. Data are means ± SD of 3 experiments