Gas chromatography mass spectrometry determination
Analysis was accomplished in the SIM mode based on the use of one target as quantification ion and two confirmation ions. PAHs were recognized according to their retention times, target and confirmation ions (
Table 1). The quantification was based on the peak area ratio of the targets to that of internal standard.In this study, the injector temperature was retained at 280°C and injection was performed in the splitless mode. The initial oven temperature was maintained at 60°C for 0.5 min, increased to 230°C at a ramp rate of 3°C min
−1 and kept for 0.5 min, then increased to 290°C at 5°C min−1 and hold for 10 min at the final temperature. Data acquisition was delayed for 12 min. The ionization was performed in ion source with electron impact mode (70ev). The ion source and triple quadrupole mass analyser temperature were kept at 230 and 280°C, respectively. A mass range of m/z 50-500 was scanned to find the retention time and diagnostic ions (quntification and confirmation ions) of the analytes. Retention time and mass spectrum of each of the standards were used to identify and confirm them. For quantitative determination, PAH standards and samples were analyzed in selected ion monitoring (SIM) mode. The retention time, diagnostic ions and quantification ion for each analyte are presented in
Table1.
| Retention time (min) | Diagnostic ions (m/z)
| PAHs |
|---|
| Quantification ion | Confirmation ions m/z (%) |
|---|
| 18.203 | 128 | 127(44.8), 129(33.5), 102(22.5) | Naphthalene |
| 30.846 | 153 | 154(93.2), 152(53.1), 151(24.6) | Acenaphthene |
| 34.790 | 166 | 165(94.0), 167(17.6), 139(10.5) | Fluorene |
| 42.151 | 178 | 179(18.8), 176(23.7), 152(13.5) | Phenanthrene |
| 42.292 | 188 | 189( 16.7), 187(13.9), 160(11.1) | Anthracene-d10 |
| 42.392 | 178 | 179(19.2), 176(24.5), 152(12.2) | Anthracene |
| 52.786 | 202 | 203(20.3), 200(22.1), 101(21.8) | Pyrene |
| 59.971 | 228 | 226(28.4), 229(22.6), 227(10.7) | Benz[a]anthracene |
| 60.217 | 228 | 226(31.3), 229(22.6), 113(23.9) | Chrysene |
| 68.106 | 252 | 253(68.6), 250(28.1), 126(55.6) | Benzo[b]fluoranthene |
| 68.176 | 252 | 253(24.4), 250(23.5), 126(23.9) | Benzo[k]fluoranthene |
| 68.634 | 252 | 253(27.4), 250(24.1), 126(21.1) | Benzo [a]pyrene |
| 75.396 | 278 | 276(29.3), 138(32.8), 139(32.5) | Dibenz[a,h]anthracene |
| 76.699 | 276 | 277(25.1), 274(23.1), 138(43.0) | Benzo[g,h,i]perylene |
Method validation
According to the calibration graphs, the linearity for Naphthalene, Acenaphthene, Flourene and for Phenanthrene, Anthracene, Pyrene, Benz[a]anthracene, Chrysene, Benzo[b]fluoranthene, Benzo[k]fluoranthene, Benzo[a]pyrene, Dibenz[a,h]anthracene, Benzo[ghi]perylene were observed over the concentration range of 0.1-2.8 and 0.35-2.8 ngmL-1, respectively. The coefficients of determination (r2) were between 0.983 and 0.999 for all PAHs. It shows that the extraction process and analytical method after validation have enough efficiency for the determination of PAHs at trace levels. In the present study, we used spiked calibration standard approach to overcome the problems caused by the matrix. In this approach, calibration standards are prepared by the addition of standard solution to blank water samples that are subjected to the same sample preparation procedure which is intended to be used for unknown samples. In this way, the standard sample matrices will have the same composition as the unknown samples and therefore the effect of matrix is reflected in both standards and unknown samples. The calibration curve is constructed using these spiked calibration standards and it is easily used to calculate the concentration of analyte (s) in unknown sample without being concerned about the matrix effects. The developed method has the advantage of using spiked calibration curves that minimize the matrix interferences.
Limits of detection (LODs) and Limits of quantification (LOQs) were calculated based on the signal-to-noise ratio of equal to 3 and 10, respectively. Recovery, repeatability, RSD%, R
2, LODs, LOQs and HORRAT of the method under optimized conditions are summarized in tables 2 and 3. As shown in these tables, all of the validation parameters examined were in compliance with EU provision No. 836/2011 (
47). Therefore, the attained validation parameters are acceptable and the optimized and validated method can be used to analyze the real samples. Hence, the optimized method were applied to analyze 50 mineral water samples which were collected from Tehran market.
| HORRATb | AverageCV(%) | Average Recovery(%) | Spiking level (ngmL-1)
| PAHs |
|---|
2.5
| 1.0
| 0.4
|
|---|
| CV (%) | Recovery (%) | CV (%) | Recovery (%) | CV (%) | Recovery (%) |
|---|
| 0.5 | 11 | 71 | 12 | 73 | 6 | 87 | 16 | 52 | Naphthalene |
| 0.4 | 9 | 81 | 12 | 77 | 6 | 94 | 8 | 72 | Acenaphthenen |
| 0.2 | 5 | 81 | 7 | 80 | 4 | 95 | 4 | 68 | Flourene |
| 0.2 | 5 | 79 | 10 | 80 | 3 | 96 | 4 | 62 | Phenanthrene |
| 0.2 | 5 | 84 | 4 | 82 | 5 | 97 | 5 | 74 | Anthracene |
| 0.5 | 11 | 65 | 7 | 59 | 8 | 72 | 18 | 65 | Pyrene |
| 0.4 | 8 | 85 | 5 | 66 | 9 | 85 | 11 | 103 | Benzo[a]anthracene |
| 0.3 | 6 | 72 | 4 | 60 | 11 | 78 | 4 | 79 | chrysene |
| 0.4 | 8 | 84 | 6 | 55 | 10 | 82 | 6 | 116 | Benzo[b]fluoranthene |
| 0.2 | 4 | 88 | 6 | 84 | 3 | 94 | 5 | 86 | Benzo[k]fluoranthene |
| 0.4 | 9 | 83 | 8 | 58 | 11 | 82 | 9 | 109 | Benzo[a]pyrene |
| 0.8 | 17 | 90 | 17 | 63 | 20 | 104 | 14 | 102 | Dibenz[a,h]anthracene |
| 0.5 | 10 | 71 | 6 | 55 | 10 | 77 | 15 | 80 | Benzo[ghi]perylene |
[object Object]
[object Object]
| Regression equation | LRe (ngmL-1) | EFd | r2 | LODc (ngmL-1) | LOQb (ngmL-1) | PAHs |
|---|
| y= 2.507 +0.6869 | 0.10-2.80 | 101.40 | 0.992 | 0.03 | 0.10 | Naphthalene |
| y=1.3637 +0.0954 | 0.10-2.80 | 115.78 | 0.995 | 0.03 | 0.10 | Acenaphthene |
| y=1.3686 +0.1663 | 0.10-2.80 | 115.71 | 0.997 | 0.03 | 0.10 | Flourene |
| y=3.6569 +0.7157 | 0.35-2.80 | 112.86 | 0.995 | 0.10 | 0.35 | Phenanthrene |
| y=3.1554 +0.3356 | 0.35-2.80 | 120.00 | 0.996 | 0.10 | 0.35 | Anthracene |
| y=1.4566 +0.0772 | 0.35-2.80 | 92.86 | 0.996 | 0.10 | 0.35 | Pyrene |
| y=0.7353 -0.0828 | 0.35-2.80 | 121.43 | 0.987 | 0.10 | 0.35 | Benzo[a]anthracene |
| y=0.8012 +0.0014 | 0.35-2.80 | 102.86 | 0.995 | 0.10 | 0.35 | chrysene |
| y=0.5118 -0.1270 | 0.35-2.80 | 120.00 | 0.983 | 0.10 | 0.35 | Benzo[b]fluoranthene |
| y=0.6653 -0.0660 | 0.35-2.80 | 125.71 | 0.996 | 0.10 | 0.35 | Benzo[k]fluoranthene |
| y=0.3071 -0.0501 | 0.35-2.80 | 118.57 | 0.993 | 0.10 | 0.35 | Benzo[a]pyrene |
| y=0.0389 +0.0038 | 0.35-2.80 | 128.57 | 0.999 | 0.10 | 0.35 | Dibenz[a,h]anthracene |
| y=0.0705 -0.0034 | 0.35-2.80 | 101.43 | 0.999 | 0.10 | 0.35 | Benzo[ghi]perylene |
[object Object]
[object Object]
[object Object]
[object Object]
[object Object]
Dispersive liquid-liquid microextraction optimization
The recovery rate is an important indicator at the extraction process. It was usually used to evaluate the efficiency of the DLLME procedure. There are many parameters that affect the recovery. In the present study, the effect of different factors such as the type and volume of extraction and disperser solvent, rpm and time of centrifuge on the recovery were evaluated and optimized. To evaluate the effect of these parameters on the recovery, we used 10.00 mL of ultrapure water containing 1.00 ngmL-1 of each PAH and 0.70 ngmL-1 of antracene-d10 as internal standard. All the optimization trials were performed three times.
The kind of extraction solvent
In order to have efficient extraction, the selection of a suitable extraction solvent is essential. The extraction solvent must have some characteristics including: low solubility in water, low volatility, High affinity to extract the desired compounds and the ability to form a clear and detectible peak in the chromatogram (
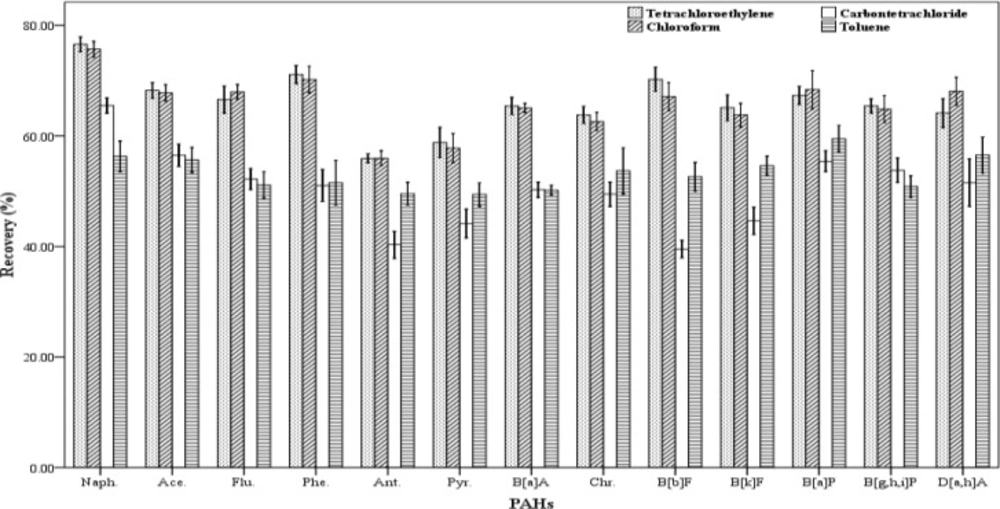
18). In this study, different organic solvents were used as extraction solvent with higher or lower density than water. The solvents including tetrachloroethylene, toluene, carbon tetrachloride, chloroform and n-hexane were examined and their ability to extract the analytes were assessed (
Figure 1). In order to evaluate the extraction efficiency of each of the organic solvents, three spiked samples were prepared. The extraction and centrifugation processes were fulfilled according to the stated method. But instead of 500 µL of each extraction solvent, 250 µL was used. The recovery values for the 13 studied PAHs are shown in
Figure 1. The results indicated that chloroform and tetra chloroethylene have higher extraction efficiency than the others. The extraction efficiencies attained for chloroform and tetra chloroethylene are similar and there were not any significant differences in recovery values between them. But a number of compounds did not result in normal peaks when tetra chloroethylene was used to extract them. Therefore, chloroform was chosen as the optimum extraction solvent.
Effect of the kind of extractant (n = 4) on the recovery of PAHs. Extraction conditions
Extraction solvent volume
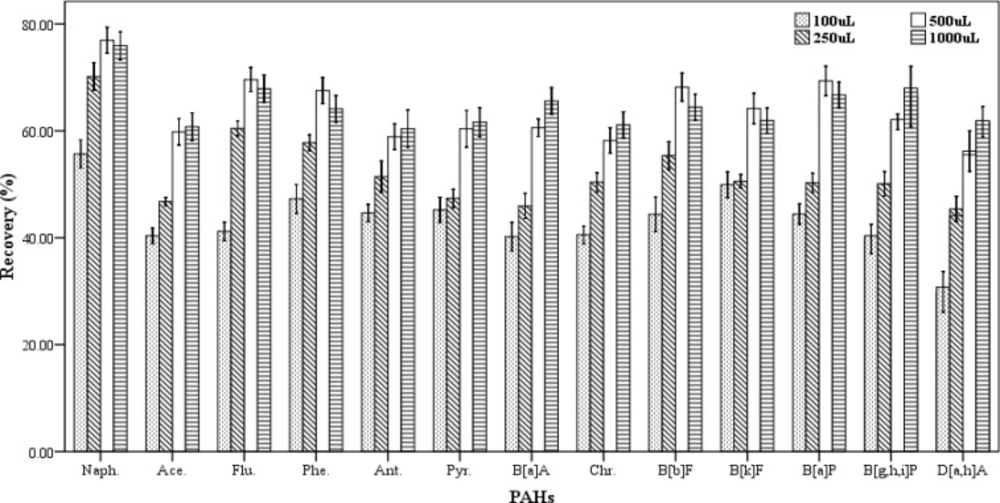
To evaluate the effect of extraction solvent volume on the recovery values of the studied analytes, different volumes of chloroform (100, 250, 500 and 1000 µL) were examined. In this study, three spiked samples were prepared for each of the volumes of the extracting solvent. At all the experiments a constant volume of 1000 µL of the dispersive solvent was applied. The extraction and centrifugation processes were done according to procedure described in section 2.3. As shown in
Figure 2, the recovery values of PAHs increase gradually with rising the volume of the extracting solvent in the range of 100-500 µL.
Figure 2 shows that the volumes of 500 and 1000 µL of the extraction solvent have the highest extraction efficiencies but there is not any significant differences in extraction efficiency between them. The results had no repeatability in case of the volumes less than 100 µL. Therefore, the volume of 500 µL of chloroform was chosen as the best volume of extraction solvent.
Effect of the extraction solvent volume on the recovery of PAHs. Extraction conditions: extraction method, DLLME procedure, ultrapure water sample volume, 10 mL; spiking level, 2.8 ngmL-1; disperser solvent volume, 1000 µL; extraction solvent, chloroform; centrifugation, 3 min., 2500 rpm, room temperature
The kind of disperser solvent
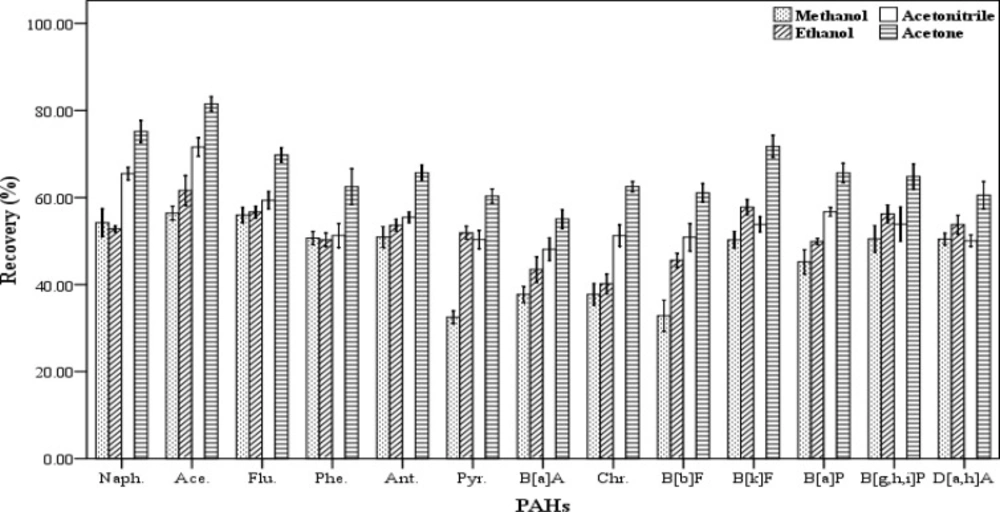
The selection of an appropriate disperser solvent is very important for the efficient extraction. The disperser solvent must have some characteristics including: the ability to be distributed in aqueous samples in order to form very small droplets and increase the surface area in contact between the analytes and the extracting solvent and solubility in the aqueous samples and the organic extraction solvents. In addition, the compound should also be not expensive and highly toxic. Therefore, the ability to form a cloudy solution or an effective emulsion depends significantly on the amount and type of the disperser solvents. In this work, acetone, acetonitrile, methanol and ethanol were tested and their influence on the recovery values were evaluated (
Figure 3). For this purpose, five spiked samples were supplied for each of disperser solvent. Then each of them was extracted by using a combination of 500 µL of chloroform as the extracting solvent and 1000 µL of each of the mentioned disperser solvents. The extraction and centrifugation processes were accomplished based on the aforesaid procedure in section 2.3. All the lower phase was collected and dried, then re-dissolved in 70 µL of chloroform. The results of these experiments are presented in
Figure 3. Maximum recovery values obtained for all 13 PAHs when acetone was used as disperser solvent compared to the others. Therefore, acetone was selected and used as disperser solvent in this study.
Effect of different disperser solvent volume (n = 4) on the recovery of PAHs. Extraction conditions: extraction method, DLLME procedure, ultrapure water sample volume, 10 mL; spiking level, 2.8 ngmL-1; disperser solvent volume, 1000 µL; extraction solvent volume (chloroform), 500 µL; centrifugation, 3 min., 2500 rpm, room temperature
Dispersive solvent volume
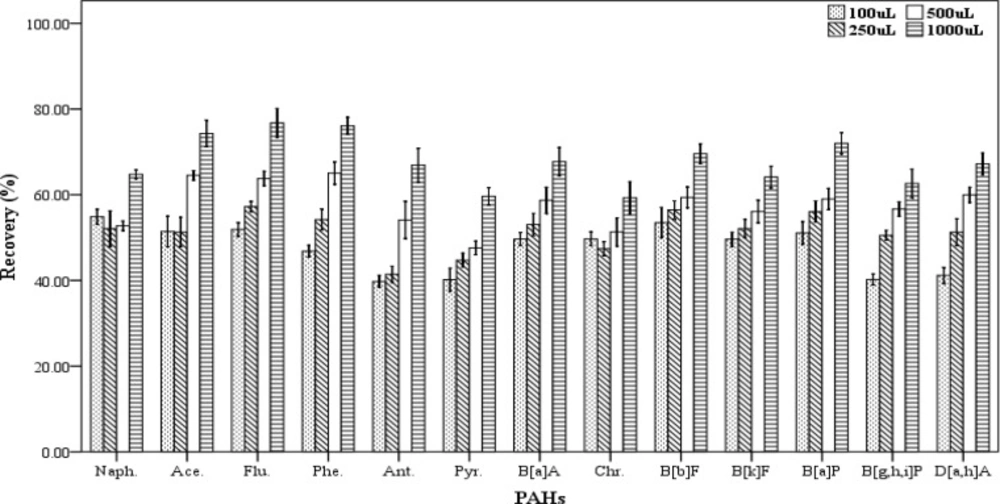
In DLLME procedure, the dispersive solvent volume is a critical parameter and its effect should be determined on the efficiency of extraction. For examining the effect of this factor on the recovery values of the 13 PAHs, five spiked samples were prepared for each of the dispersive solvent volume. In this work, the different volumes (100, 250, 500 and 1000 µL) of acetone as dispersive solvent containing 500 µL chloroform as the extracting solvent were applied for extraction. The extraction and centrifugation processes were accomplished based on the aforementioned technique in Section 2.3. The settled phase was collected completely and dried, then re-dissolved in 70 µL of chloroform. As shown in
Figure 4, increasing the dispersive solvent volume results gradually in increased recovery values of PAHs. The highest efficiency of extraction was achieved when 1000 µL of acetone was used. Then, a mixture of the extracting and dispersve solvents with volumes of 500 and 1000 µL, respectively were selected as the optimum volumes for further trials.
Effect of the disperser solvent volume (n = 4) on the recovery of PAHs. Extraction conditions: extraction method, DLLME procedure, ultrapure water sample volume, 10 mL; spiking level, 2.8 ngmL-1; disperser solvent, acetone, extraction solvent volume, 500 µL; centrifugation, 3 min., 2500 rpm, room temperature
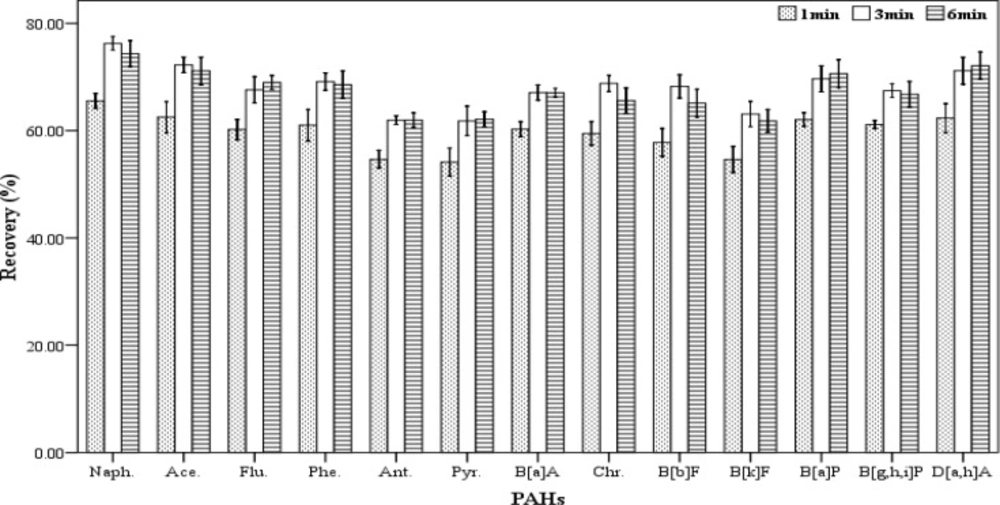
Centrifugation time
In order to assess the effect of centrifugation time on the extracting efficiency, a range of centrifugation times (1, 3 and 6 min) were tried. For each of the mentioned times, five spiked samples were prepared. For extraction, a mixture containing 500 µL chloroform and 1000 µL acetone were used as the extracting and disperser solvents, respectively. The extraction process was accomplished according to the described procedure. All of the experiments were performed at 5000 rpm. The results (recovery values) are shown in
Figure 5. According to the results, the centrifugation times 3 and 6 minutes have the highest effect on the amount of recovery but there was not a significant difference between them. Therefore, 3 min was applied as the optimum centrifugation time for further tests.
Effect of centrifugation time (n = 3) on the recovery of PAHs. Extraction conditions: extraction method, DLLME procedure; ultrapure water sample volume, 10 mL; spiking level, 2.8 ngmL-1; disperser solvent (acetone) volume, 1000 µL; extraction solvent (chloroform) volume, 500 µL; centrifugation rpm, 2500, room temperature
Analysis of real samples
Khalili Zanjani MR,
et al. (
26) and Rezaee M,
et al. (
38) have reported the use of DLLME method along with GC-FID for the analysis of polycyclic aromatic hydrocarbons.
Khalili Zanjani MR, et al. have used 1-undecanol as the extraction solvent while Rezaee M, et al., have used tetra chloroethylene for extraction.
In the present study, chloroform was used as the extraction solvent and mass detector was used coupled to the gas chromatograph.
Satisfactory figures of merit were obtained. The optimized and validated procedure was used to determine PAHs in 50 real water samples (bottled mineral water). No treatment was conducted such as filtration on the samples, prior to extraction process. Extraction process, centrifugation and analysis were accomplished according to the described procedure. A blank and three spiked ultrapure water at concentration of 1 ngmL
-1 were used beside the real water samples, per working day. The results of the samples analysis show that the concentrations of PAHs in the real samples (mineral water samples) were lower than the LOQ in all the samples. There are only few published papers for determination of PAH in mineral water. In one of these studies, Humood F. Al-Mudhafa
et al. (
48) analyzed twenty-five bottled water brands for extractable semivolatiles (SVs), including PAHs, listed in the US-EPA 525.2 method; and found that all samples were free of SVs contamination. Our results are in agreement with the above mentioned study.
But in some studies, some contamination have been found in mineral water. For example, In one of these studies, Ma Teresa Pena
et al. (
49) determined eighteen PAHs in drinking water samples (tap, bottled, fountain, well) and reported sum of their concentration between 127.8 ngL
−1 and 413.2 ngL
−1. In another study, Albert Guart
et al. (
50) detected PAHs in 18 out of 77 bottled water samples at levels between 0.005 and 0.202 μgL
-1. They found that the most detected compound was naphthalene, which was detected in 16 samples at concentrations of 0.005-0.202 μgL
-1. Also, CuneytGuler, (
51) found that a significant number of bottled waters in Turkey contained some elements (
e.g. sodium, chloride, sulfide, fluoride, PAHs and several heavy metals) above the maximum concentration allowed for bottled waters by the Turkish legislation.
Considering the few studies regarding PAHs contamination in drinking water in Iran, to get a clear picture of contamination of drinking water, comprehensive monitoring of PAHs in water in different provinces and seasons are suggested.