Electrochemical behavior of INH on PEDOT modified Au-electrode
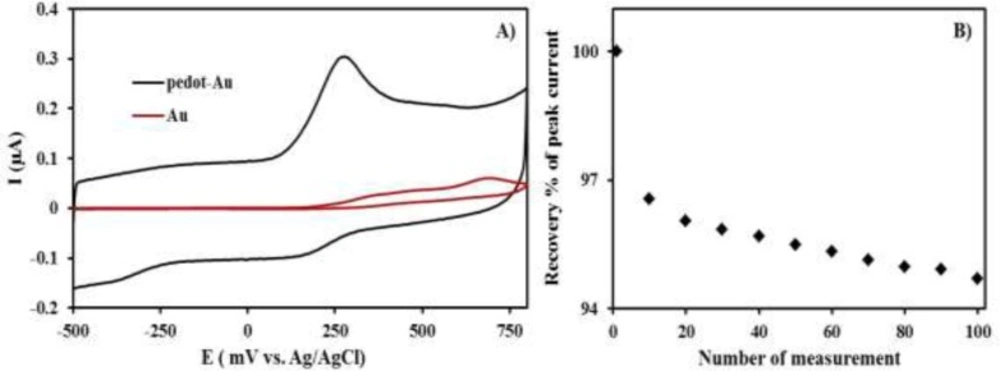
Cyclic voltammetric curves (CVs) data were obtained on bare gold electrode and PEDOT-modified gold electrode in phosphate buffer solution (pH: 9.2), containing 1 µM INH, as shown in
Figure 1. A single oxidation peak was recorded at 0.25 V, in accordance with the CV of INH, in this solution (curve blue). A relatively similar curve was received for the bare gold electrode, in terms of CV in the same solution (curve red), except that the CV had a smaller current and higher INH oxidation potential. As seen in
Figure 1A, there is a significant difference in terms of intensity of the peak current densities, and that of the PEDOT-Au electrode was approximately 4-folds higher than that of the bare Au. The higher peak current densities for the PEDOT/Au electrode lead to more favorable oxidation of INH than of the bare Au electrode. Thus, the higher peak currents for INH oxidation on the PEDOT/Au electrode could be more advantageous, because of its property of higher effective surface areas, improved mass transport, and higher catalytic effect due to electronic properties, compared to the other conductive.
A) CVs of bare gold electrode and PEDOT modified gold electrode in an aqueous solution (pH: 9.2) containing 1 µM INH, B) The curve of the stability data for the PEDOT modified Au-electrode
Response stability and reproducibility of the PEDOT modified Au-electrode
Response stability of PEDOT/Au electrode was evaluated by measuring the anodic peak current response at a fixed 1 µM in phosphate buffer solution (pH: 9.2) at the identical surface of PEDOT/Au electrode without renewal for 20, 50 and 100 times in a day. 97%, 96%, and 95% of the initial amperometric response (the mean peak current for 1 µM INH was 0.31 in
Figure 1A) was obtained as can be seen in,
Figure 1B, respectively, indicating PEDOT/Au has shown good stability under continuous use. In addition to this, renewed surface of PEDOT/Au electrode in phosphate buffer solution (pH: 9.2) was evaluated in terms of its reproducibility. The average peak current for 1 µM isoniazid was 0.307 0.005 (n=6). This data definitely showed that this modified electrode has a remarkable reproducibility. However, PEDOT/Au electrode was prepared only one time. Despite of its high reproducibility, this electrode was used during the whole analysis due to its response stability.
Optimization of Experimental Parameters by Central Composite Design (CCD)
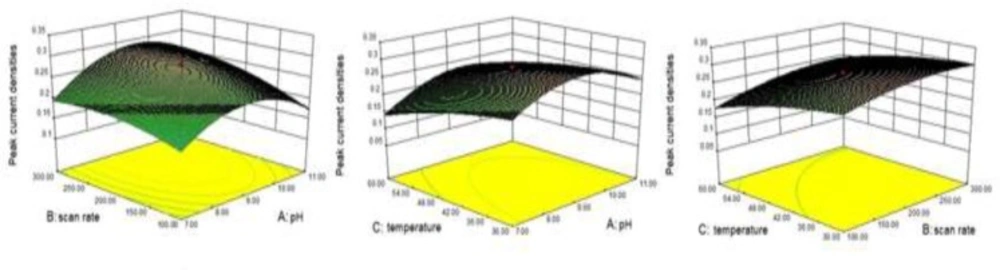
A CCD was performed to optimize and determine of the interaction between selected factors the operating parameters for determining INH levels in pharmaceuticals. Scan rate (mV/s, B), pH (A) and temperature (C, C) are the three significant selected factors necessary to carry out CCD. In order to make a decision regarding the significance of these three parameters, preliminary experiments and knowledge obtained from previous studies were utilized. The range of the three factors were determined as follows: pH (7-11), scan rate (100-300 mV/s), and temperature (30-60 C). The response variable of the CCD was accepted as being the peak current densities of INH. The CCD method was carried out by using eight factorial and six axial runs, and, in addition to these measurements, the model was maintained in six axial runs at center points. In order to carry out rotatability, 1.689 (α = (23)1/4 ∼1.689) value was selected to be the α value of the CCD.
The experimental conditions for CCD and the results of peak current densities are summarized in
Table 1. Three-dimensional graph were plotted, which is called as response surface, for the experimental results of the obtained peak current densities from CCD
Surface plot for INH peak current densities A) Effect of A and B and their reciprocal interaction B) Effect of A and C and their reciprocal interaction C) Effect of B and C and their reciprocal interaction
(
Figure 2), and fitted the experimental data to the response surface in order to obtain the optimum conditions. The following equation was improved for the quadratic regression model in coded terms:
Y = 0.28+ 0.017A +0.027 B -0.035 C+0.014 AB+0.011 AC+0.00625BC- 0.061 A2 - 0.018 B2- 0.013 C2
| Experiment | A: pH | B: Scan Rate (mV/s) | C: Temperature (C) | IF: Peak Current densities |
|---|
| 1 | 11.0 | 100 | 30.0 | 0.18 |
| 2 | 9.0 | 32 | 45.0 | 0.15 |
| 3 | 9.0 | 200 | 45.0 | 0.27 |
| 4 | 9.0 | 368 | 45.0 | 0.35 |
| 5 | 11.0 | 300 | 60.0 | 0.19 |
| 6 | 7.0 | 300 | 60.0 | 0.12 |
| 7 | 12.3 | 200 | 45.0 | 0.20 |
| 8 | 7.0 | 100 | 60.0 | 0.15 |
| 9 | 7.0 | 100 | 30.0 | 0.21 |
| 10 | 9.0 | 200 | 70.2 | 0.18 |
| 11 | 7.0 | 300 | 30.0 | 0.2 |
| 12 | 5.6 | 200 | 45.0 | 0.06 |
| 13 | 9.0 | 200 | 19.8 | 0.35 |
| 14 | 11.0 | 100 | 60.0 | 0.12 |
| 15-20 | 9.0 | 200 | 45.0 | 0.27-0.28 |
Where Y is the response factor corresponding to the peak current densities, and A, B, and C represent the three significant factors mentioned above. The p value was 0.0235, in accordance with the statistical parameters obtained from the analysis of variance for the model. This model could consider being significant, due to a p value of < 0.05, and it was claimed that pH and temperature significantly affected the peak current densities of INH. Maximum desirability was observed on the coordinates of a pH of 9.2, a scan rate of 260 mV/s, and a temperature of 30 C. To confirm the point prediction values, experiments (n = 10) were maintained to determine the response peak current densities values, using the conditions recommended by the model. The mean peak current density value of the results was 0.315 ± 0.037, indicated a fairly good agreement with the predicted results.
Validation and Quantitation
The proposed method was validated by evaluating the parameters of specificity, linearity, sensitivity, precision, accuracy, recovery, and stability, in accordance with International Conference on Harmonization Q2B guidance (
38).
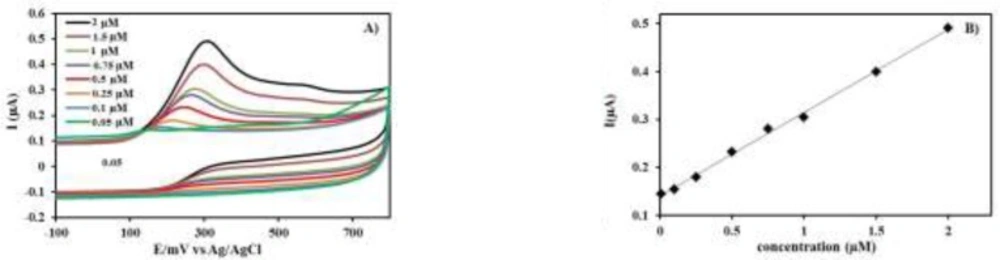
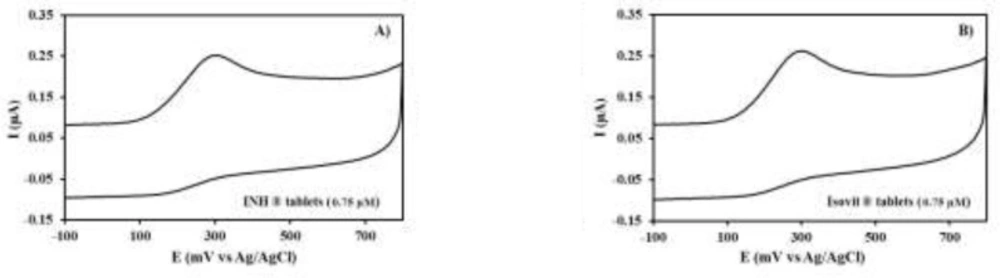
The specificity of the proposed method was clearly evidenced by the identical oxidation peak of INH in standard solution (
Figure 3) and drug samples containing the excipients and vitamin B6 (
Figure 4). The oxidation peaks of vitamin B6 and drug excipients were not identified at PEDOT/Au electrode under the described experimental conditions.
A) CVs as increasing concentration of INH (0.05-2 μM) in phosphate buffer solution (0.1M, pH 9.2) at PEDOT/Au electrode. B) Calibration curve for INH of different concentrations from (0.05-2 μM
CVs of drug solutions (0.75 µM) in phosphate buffer solution (0.1M, pH 9.2) at PEDOT/Au electrode. A) I.N.H tablets B) Isovit tablets
The linearity of method was shown by linear calibration curves obtained by plotting peak current densities of INH versus concentration of INH.
Figure 3A exhibits the CVs recorded under increasing concentrations of INH (0.05, 0.1, 0.25, 0.5, 0.75, 1, 1.5, and 2 μM). The obtained calibration curves are also shown (
Figure 3B). Linear regression analysis was used to evaluate the linearity, which was calculated by least-squares regression analysis. The average of six calculated linear regression equation was Y= 0.1727 C+ 0.1357. C is the concentration (μM) of INH, and Y is peak current densities of INH (μA). Furthermore, standard deviation of intercept (Sa) and slope (Sb) of regression lines from these six linear regression equations were calculated as 0.00074 and 0.0028, respectively. The correlation coefficient was 0.9998, which definitely represents a good linearity over the working concentration range.
The limit of detection (LOD) and the limit of quantitation (LOQ) were calculated as 3.3 /S and 10 /S, respectively, where σ is the standard deviation of the intercept of the regression line, and S is the slope of the regression line of the calibration curve. The LOD and LOQ were 0.014 μM and 0.043 μM, respectively. The LOD value is sufficient for INH analysis in the pharmaceutical preparations.
In order to assess the precision of the methods, the percent relative standard deviation (RSD %) and the accuracy of method relative error (RE) values were evaluated with intra-day and inter-day measurement at three different concentration levels of INH that are known to be quality control solutions (0.1, 0.75 and 1.5 µM). The RSD % values for intra-day and inter-day precision of proposed methods were ≤ 7.97 %, and the RE values for the intra-day and inter-day accuracy studies of method was between -5.65 and 4.03. These results are shown in
Table 2.
| Added(M) | Intra-day
| Inter-day
|
|---|
| Found SDa(M) | RE | RSD % | Found SDa(M) | RE | RSD % |
|---|
| 0.1 | 0.104 ± 0.005 | 4.03 | 5.47 | 0.099 ± 0.008 | -0.79 | 7.97 |
| 0.75 | 0.742 ± 0.020 | -0.98 | 2.72 | 0.707 ± 0.047 | -5.65 | 6.66 |
| 1.5 | 1.493 ± 0.018 | -0.44 | 1.18 | 1.517 ± 0.049 | 1.19 | 3.23 |
: Average of ten determinations, RSD %: Relative standard deviation. RE: Relative error.
Whether excipients and other active drug substance (Vitamin B6) interfered the analyte in the tablets or not was determined by recovery test which based on addition of known amounts of pure drug to pre-analyzed formulations of INH. Recovery study (R %) was carried out by spiking known quantities (0.1, 0.75, and 1.5 µM) of standard into the drug solutions, which were prepared from Isovit® and INH® tablets. The analytical recovery was calculated by the following formula:
R % = [Ct-Ce/Ca]x100
where Ct is the total concentration of INH in the added samples, Ce is the concentration of INH to be estimated in pharmaceutical tablets, and Ca is the concentration of standard INH added. The obtained results had good accuracy for reliable electro-analytical analysis of INH, with a mean recovery of 99.8 4.36 % for Isovit® tablets, and 99.1 3.27 % for INH® tablets.
Solution stability of standards and tablet samples were assessed at room temperature and 5C. For this, standards and tablet solutions at 0.1, 0.75 and 1.5 µM were analyzed initially and at different time intervals (6, 24 and 48 h). The standards and sample solutions were stable for at 24 hours at room temperature and at least 48 hours at 5 C.
The developed voltammetric method was applied to analyze the commercially available formulations of Isovit
® and INH
® tablets (
Figure 4). To evaluate the amount of INH present, the samples were analyzed 10 times after preparing the drug as mentioned previously, in the “Experimental Procedure” section. The INH content of pharmaceutical tablets were calculated on the basis of the standard calibration curve and the obtained results expressed as a percentage of drugs related to label claim was shown in
Table 3. In addition this, these results were statistically compared with the official USP methods (
17,
39), using the two one-sided equivalence test (TOST). The upper and lower acceptable limits for the difference in the amount of the active ingredient at each dosage have been predetermined as -3.0% to 3.0% (40). The critical t value for 18 degrees of freedom, with a set for the TOST at 0.05 was 1.734. The calculated confidence intervals for Isovit
® and INH
® tablets were -0.619 to 1.438, and -0.747 to 1.432, respectively. Therefore, it was concluded that the results obtained with the proposed method and the USP reference assay were equivalent.
| Drug | Label claim (mg per tablet) | Founda SD(mg) | RecoverySD, (%)
| Confidience Interval
|
|---|
| Proposed Methoda | Official U.S.P.Methods17,39 | Lower | Upper |
|---|
| Isovit | 100 | 100.2 ± 1.55 | 100.2 ± 1.55 | 99.82 ± 1.11 | -0.619 | 1.438 |
| I.N.H | 300 | 300.5 ± 5.36 | 100.2 ± 1.68 | 99.82 ± 1.11 | -0.747 | 1.432 |
: Average of ten determinations.