Materials and reagents
Reference standards of CPS, creatine and creatinine were obtained from National Institutes for Food and Drug Control (Beijing, China). The creatinine phosphate disodium salt reference standard was purchased from ChromaDexTM (California, USA). CPS raw material and injections were kindly supplied by Hainan Quanxing Pharmaceutical. Acetonitrile (ACN) was chromatographic grade and procured from Spectrum Chemical Mfg. Corp. (Gardena, USA). Analytical grade orthophosphoric acid, sodium hydroxide, potassium permanganate and chromatographic grade KH2PO4 were purchased from Chengdu Kelong Chemical Reagent Company (Chengdu, China). Chromatographic grade tetrabutylammonium hydroxide (TAH) was purchased from Tianjin Guangfu Fine Chemical Research Institute. Demineralised water (≥ 18.0 Ω cm-1) was prepared with a Millipore Milli-Q plus purification system (Bedford, USA).
Instruments
A pH meter (Mettler Toledo, Germany) and a Mettler Toledo balance were employed in the preparation of sample solutions and mobile phase. Photo stability and thermal stability studies were carried out in Multi-Drug Stability Test Chamber (YSEI, China) and DHG Series Electrothermal Constant-temperature Drying Box (SANFA, China), respectively.
An Agilent 1260 Liquid Chromatograph (Agilent, USA) consisting of a quaternary pump, online degasser, column heater, autosampler and diode array detector was employed for analysis. Data collection and analysis were performed on an Agilent OpenLAB CDS ChemStation (Rev.C.01.03, Agilent Technologies).
Chromatographic conditions
Compounds were separated on a Hypersil BDS-C18 column (250 mm × 4.6 mm I.D., 5 μm) (Dalian Elite Analytical Instruments Co., Ltd. Dalian, CHINA). The aqueous mobile phase contained 0.2% (w/v) TAH and 0.2% (w/v) monopotassium phosphate. The final pH of the mobile phase was adjusted to 6.6 by diluted orthophosphoric acid. The eluted compounds were monitored at 210 nm. Column temperature was set at 30 ˚C and the flow rate was 1.0 mL min-1. 20 μL of sample was injected to HPLC system. The mobile phase was filtered through 0.22 μm membrane filters and degassed prior to using.
Preparation of solutions
The aqueous diluent for standards and samples contained 0.2% (w/v) TAH and 0.2% (w/v) KH2PO4 (adjusted to pH 9.0 by caustic soda solution).
Preparation of standard solution
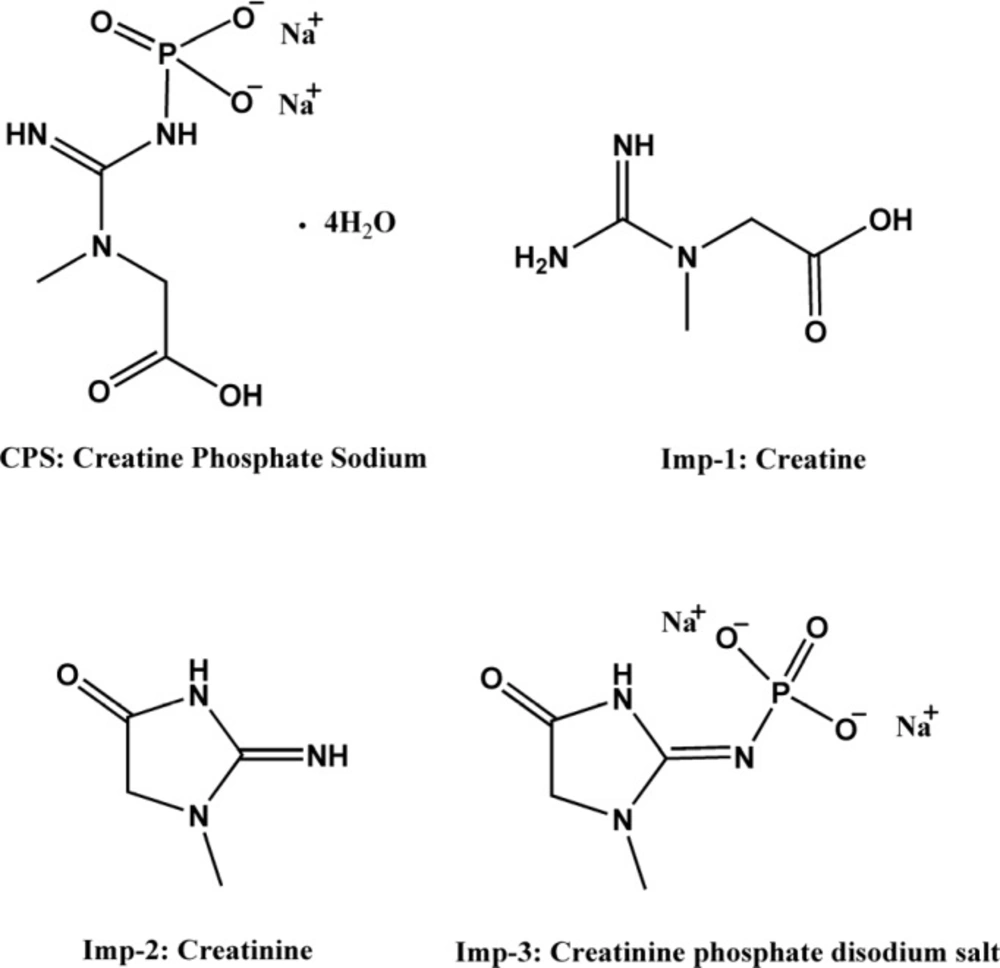
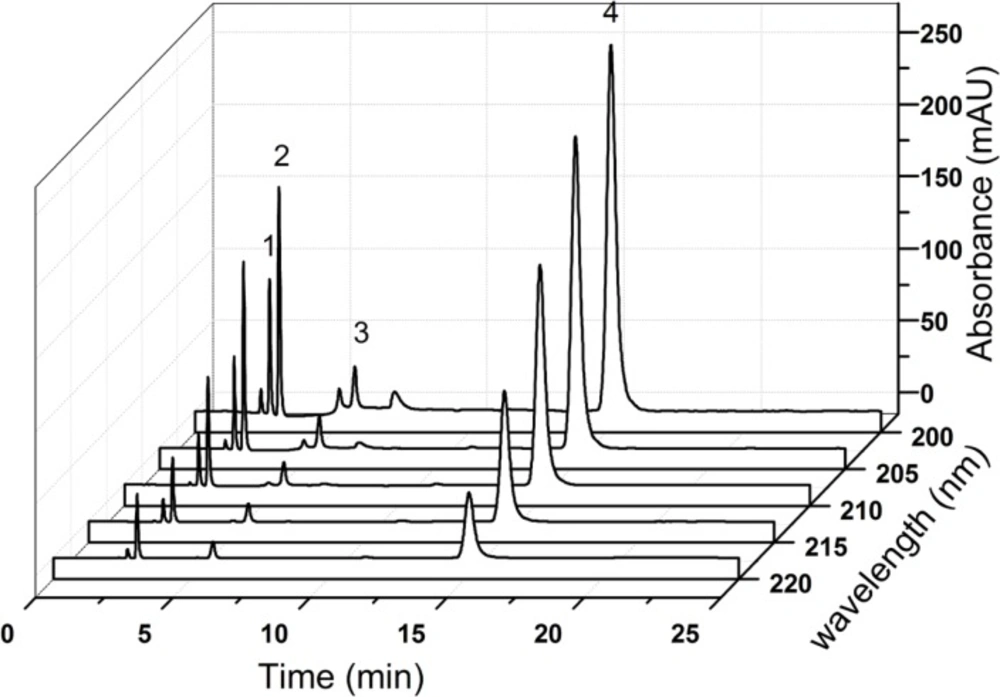
Stock solutions of the individual compounds (CPS, creatine, creatinine and creatinine phosphate disodium salt) were prepared by dissolving appropriate amount reference substance in the diluent. The mixed standard solution was prepared by diluting the respective stock solutions to the final concentrations in a 10 mL volumetric flask. The final concentration of CPS, creatinine phosphate disodium salt and other impurities were 100 μg mL-1, 2.5 μg mL-1, and 5 μg mL-1, respectively.
Preparation of sample solution
About 13.4 mg (water, 25%) of sample was accurately weighed and transferred to a 10 mL volumetric flask. A proper amount of diluent was added and shaken for 1 minute, then diluted up to the mark (1 mg mL-1). 1 mL of this solution was removed into a measuring flask of 10 mL. Then the flask was filled to full volume with the diluent to get the desired concentration (100 μg mL-1). The concentrated solution (1 mg mL-1) was for the analysis of creatine, creatinine and creatinine phosphate disodium salt, and the final solution was for the analysis of CPS.
Stress study
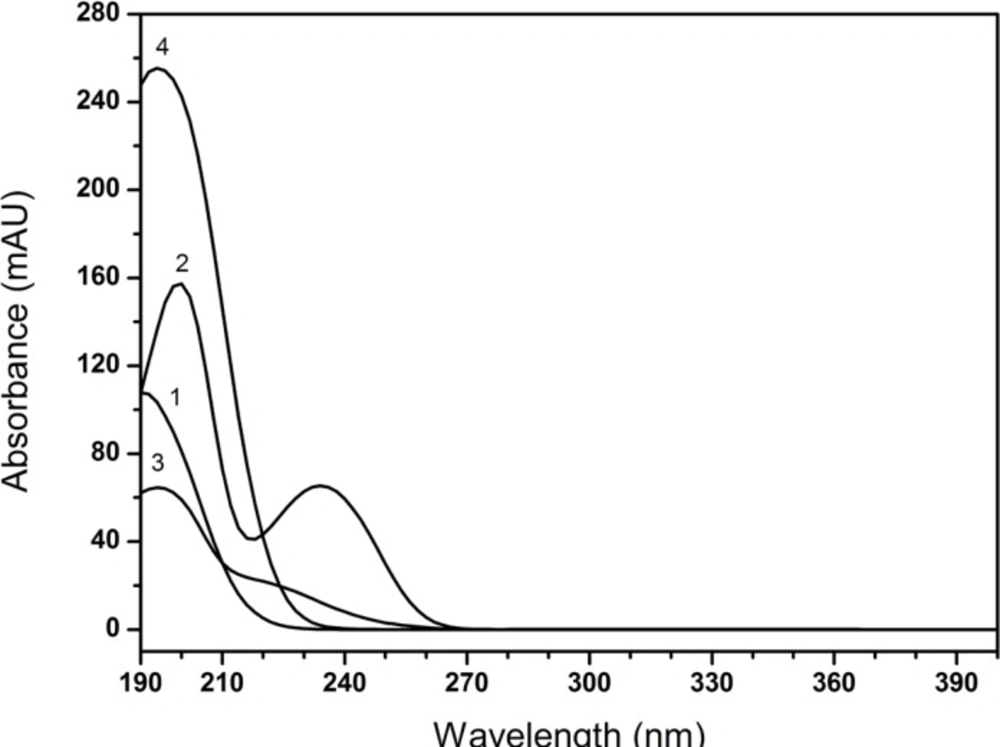
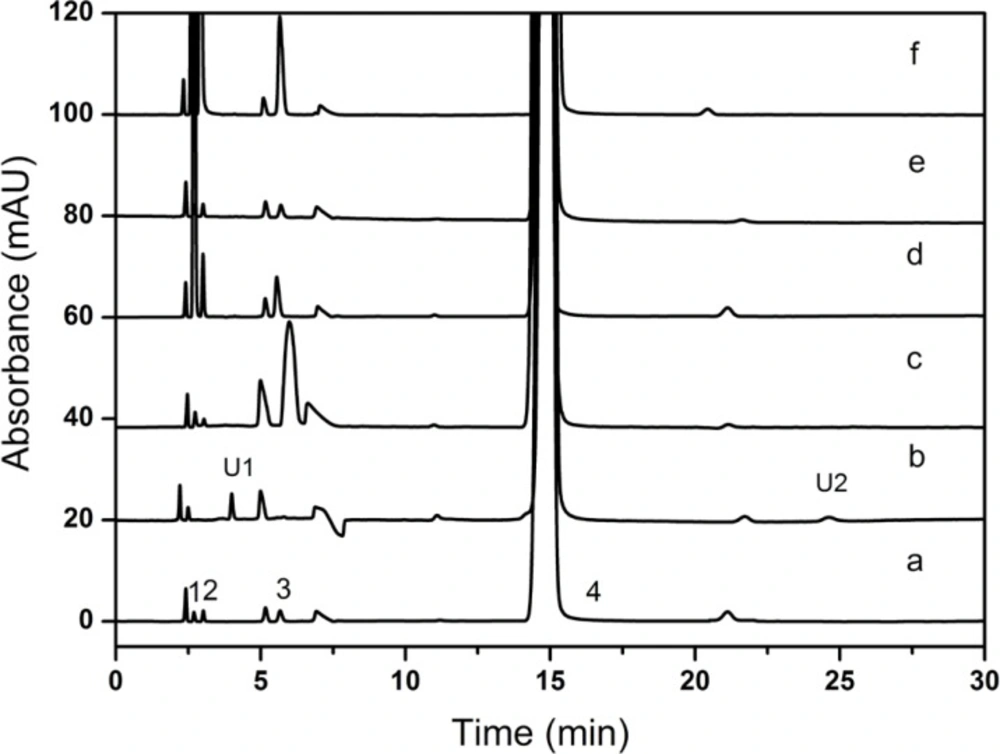
The stress conditions employed for the degradation study included base hydrolysis (1 N NaOH), acid hydrolysis (0.01 N H3PO4), thermal treatment (60 ˚C), oxidation (0.025 g mL-1 KMnO4) and photolytic condition (4500 Lx ± 50). The samples were treated for 6 h, 40 minutes, 40 minutes for base, acid hydrolysis and oxidation, respectively. Whereas for thermal and photolytic condition studies, the samples were exposed for 10 days. After degradation studies, the stress samples were neutralized. Sample solutions were prepared as represented in “Preparation of sample solution”. The CPS peak purity of the stressed samples was monitored by the PDA detector in the wavelength range of 190-400 nm.
Method development and optimization
The chromatographic conditions including mobile phase composition, pH of mobile phase and diluent, detection wavelength were investigated to obtain the satisfactory analytical performance.
Selection of the mobile phase, detection wavelength and pH
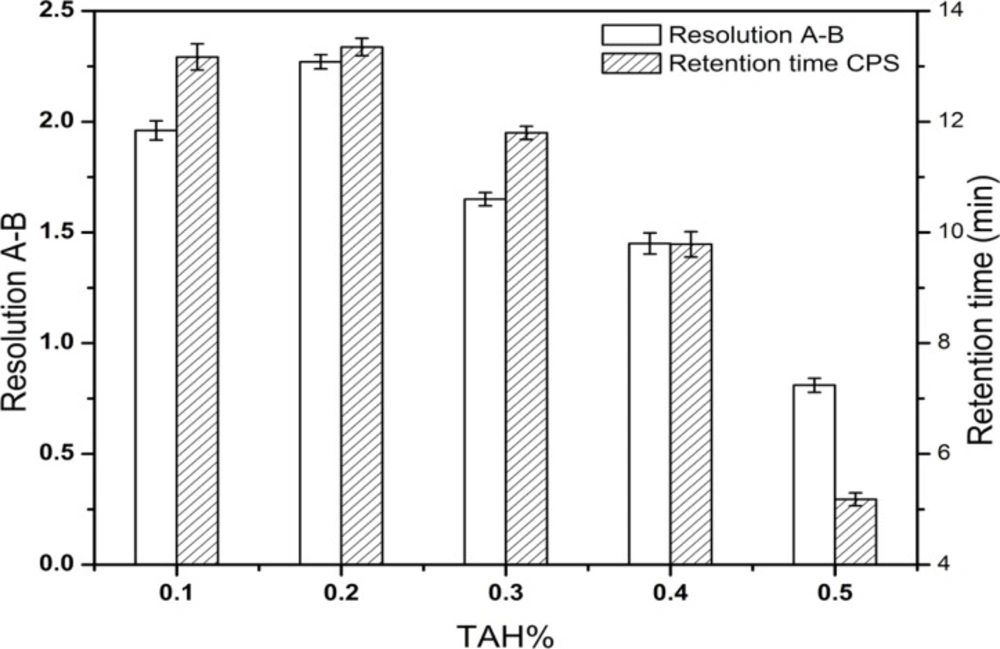
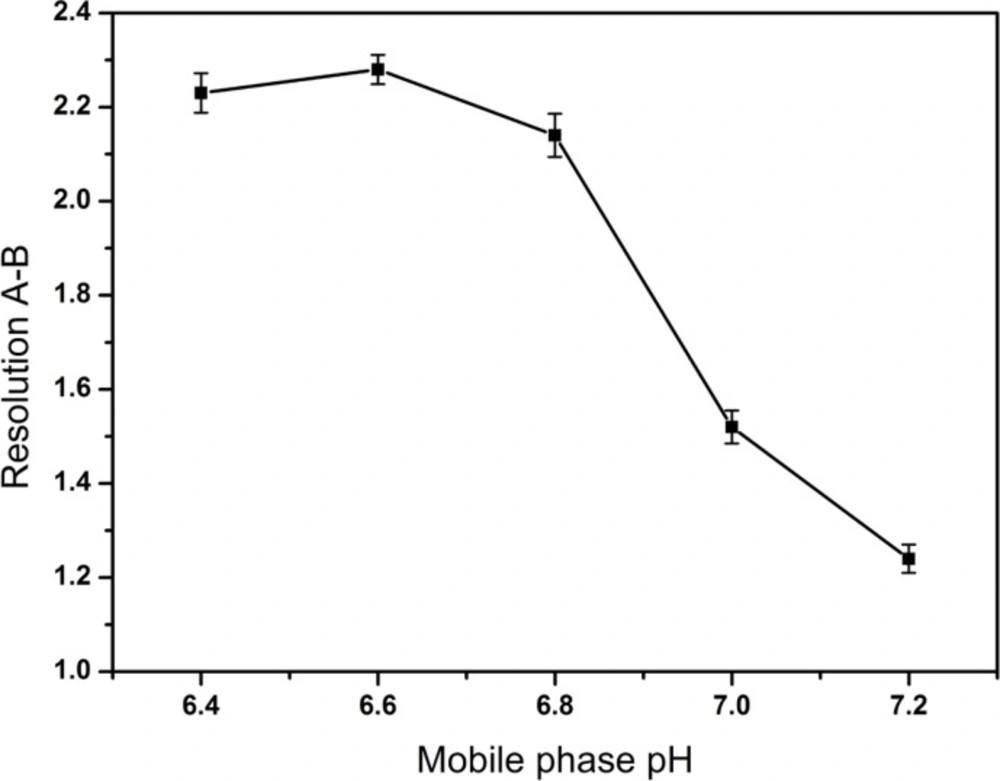
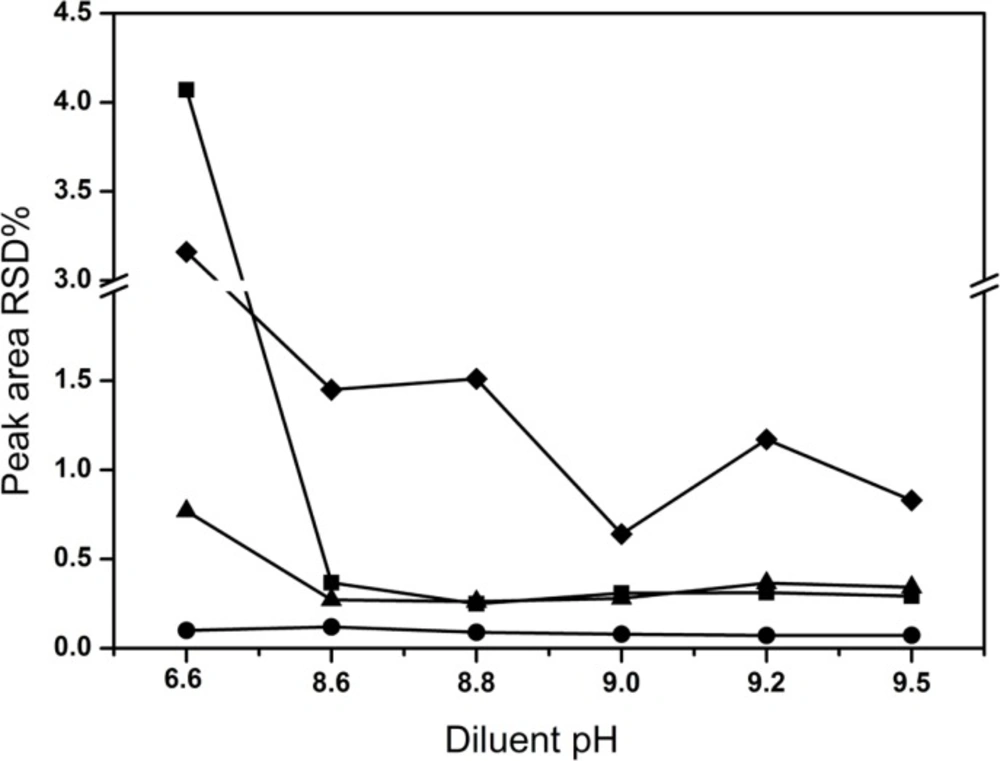
The most effective amount of TAH in the mobile phase was determined in five different concentrations (0.1, 0.2, 0.3, 0.4, 0.5%), and the most appropriate amount of KH2PO4 in mobile phase was 0.2%. The pH values of the mobile phase and the diluent were investigated in the range of 6.4-7.2 and 6.0-9.5, respectively. A suitable wavelength was required for the determination of CPS and three related substances simultaneously. The appropriate wavelength in the mobile phase was determined by wavelength scanning over the range of 190-400 nm.
Methodoptimization
The column was determined by comparing the three manufacturers of Agilent (Eclipse XDB-C18 4.6 × 250 mm, 5 μm), Elite (Hypersil BDS-C18 4.6 × 250 mm, 5 μm) and Waters (X BridgeTM Shield RP18 4.6 × 250 mm, 5 μm). The flow rate was investigated at 0.8, 1.0 and 1.2 mL min-1. The column temperature and the sample volume were also investigated in the experiment.
Method validation
System suitability
System suitability checks are used for chromatographic methods to ensure that the system is sufficiently sensitive, specific and reproducible for the current analytical run (
15). System suitability test was performed by assessing the six replicate injections of a system suitability solution. The system suitability parameters including retention time, number of theoretical plates, resolution, tailing factor, the peak area and retention time relative standard deviation (RSD, n = 6) of each compound were evaluated.
Limit of quantitation (LOQ) and limit of detection (LOD)
The LODs (S/N = 3) and LOQs (S/N = 10) for CPS, creatine, creatinine and creatinine phosphate disodium salt were estimated by injecting a series of dilute solutions with known concentrations, respectively. The precision study at the LOQ level was also carried out by injecting six individual preparations of the analytes standard solutions.
Linearity
The linearity of CPS and its related substances were studied by determining standard solutions at six different concentrations. The standard stock solution of CPS was diluted to 80-120% of the assay method concentration (100 µg mL-1). The linearity test solutions for creatine, creatinine, and creatinine phosphate disodium salt were prepared by diluting the impurity stock solutions to different concentrations, ranging from LOQ to 6 μg mL-1, and the range was approximately equivalent to 0.1-6.0% (w/w) of CPS in injections. All solutions were injected in triplicate.
Relative response factor
Relative response factor (RRF) was established for impurities 1, 2 and 3 as the ratio of slope of CPS and slope of impurities. Slope value of impurities obtained with linearity calibration plot (in the
Table 2) was used for RRF determination. Slope value of CPS obtained with linearity calibration plot at impurities concentration was used for RRF determination. The linearity of CPS at impurities concentration was evaluated at six concentrations from LOQ concentration to 120% of 5 µg mL
-1 (0.102-6.136 μg mL
-1).
| Compd. | Regression equation | R2 | Linear rangeµg·mL−1 | LOD (ng) | LOQ (ng) |
|---|
| Imp-1 | Y = 27.587 X+2.4935 | 0.9998 | 0.127~6.102 | 1.04 | 2.59 |
| Imp-2 | Y = 68.643 X+0.0293 | 0.9998 | 0.110~6.587 | 0.22 | 0.50 |
| Imp-3 | Y= 28.2999X+1.8199 | 0.9992 | 0.090~6.000 | 0.60 | 1.80 |
| CPS | Y = 38.681 X-20.514 | 0.9995 | 83.021~118.305 | 0.90 | 2.00 |
Accuracy
Accuracy was determined by the method of standard addition recovery studies. This experiment was employed by adding the known amounts of CPS, creatine, creatinine, and creatinine phosphate disodium salt of three different concentrations into the drug samples whose concentrations were known. The different concentrations of the mixed samples were prepared in triplicate.
Precision
The repeatability of the proposed method was validated by analyzing the preparations of the six QC samples. The intermediate precision of the method was also verified on different days by different analyst, different columns and different HPLC instruments in the same laboratory.
Robustness
Small and deliberate changes in HPLC parameters were made to determine the robustness of the analytical method. The HPLC parameters variation included the proportion of TAH or KH
2PO
4 (± 0.01%) in the mobile phase, pH (± 0.2 units) of the mobile phase, column temperature (± 5 ˚C), flow rate (± 0.2 mL min
-1), wavelength (± 5 nm), and column batches. The selected parameters are those most likely to be changed and tend to have potential influence on the analytical results when the method is employed in practice (
16). The drug samples were spiked with the impurity reference substances at impurity tolerance level. The effect of the HPLC parameters variation on the retention time and peak parameters were studied.
Specificity
The specificity of a method may be defined as the ability to accurately measure the analyte in the presence of all potential sample components (
17). Assays were carried out for the stress samples against a qualified reference standard. The mass balance (% assay + % of impurities + % of degradation products) was calculated for all the samples.
Solution stability
The solution stability was assessed by analyzing the sample and standard solutions which were stored at room temperature for 12 h.
Method application
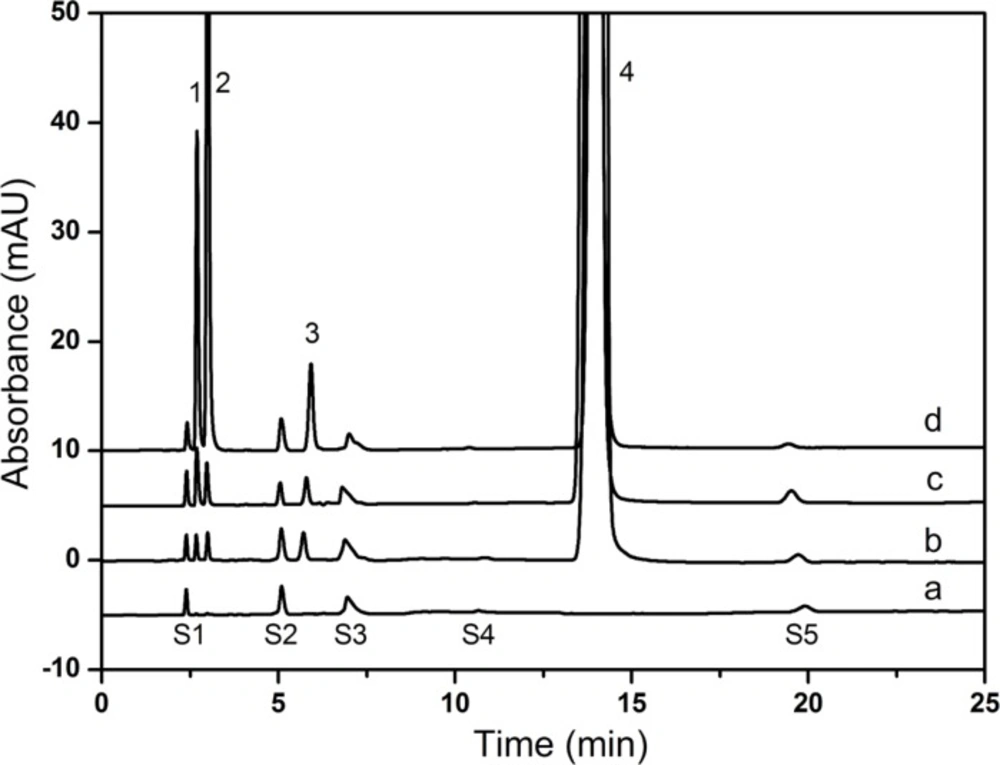
The validated HPLC method was successfully applied for the assay and related substances determination of CPS in drug substance and drug product. The drug products from three different manufacturers and the drug substance of three batches were included.