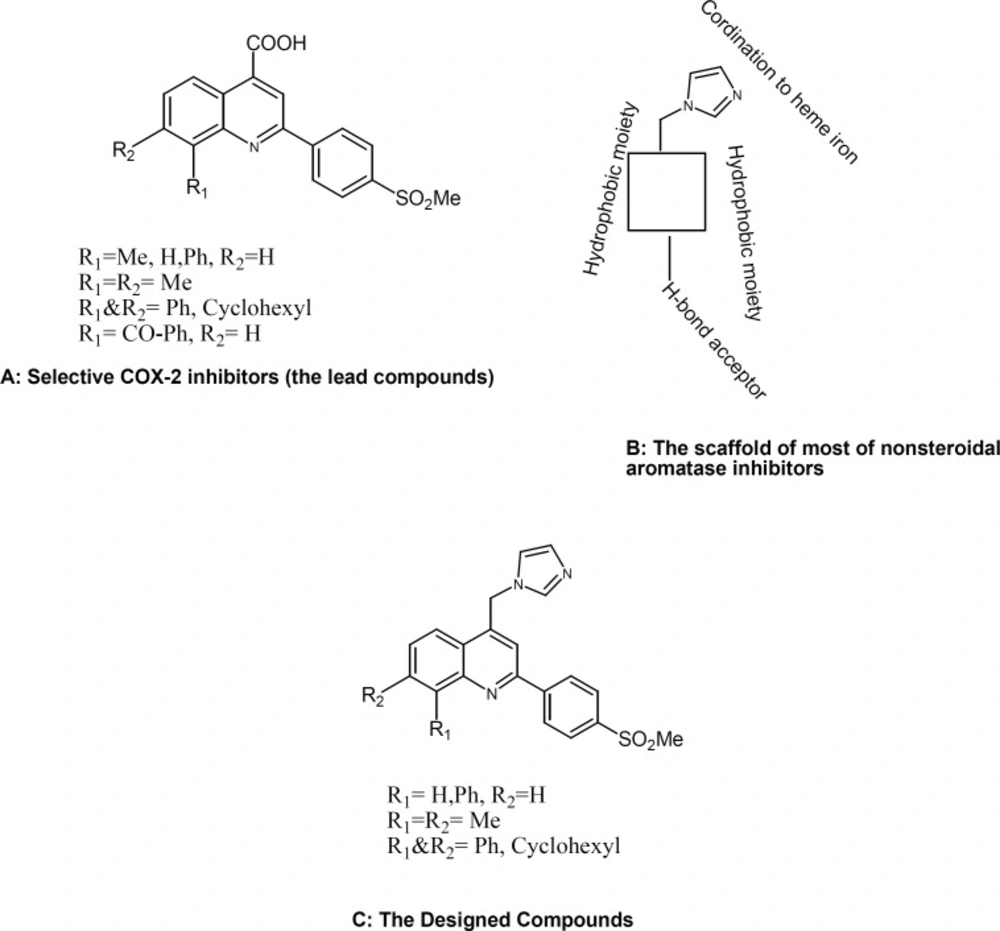
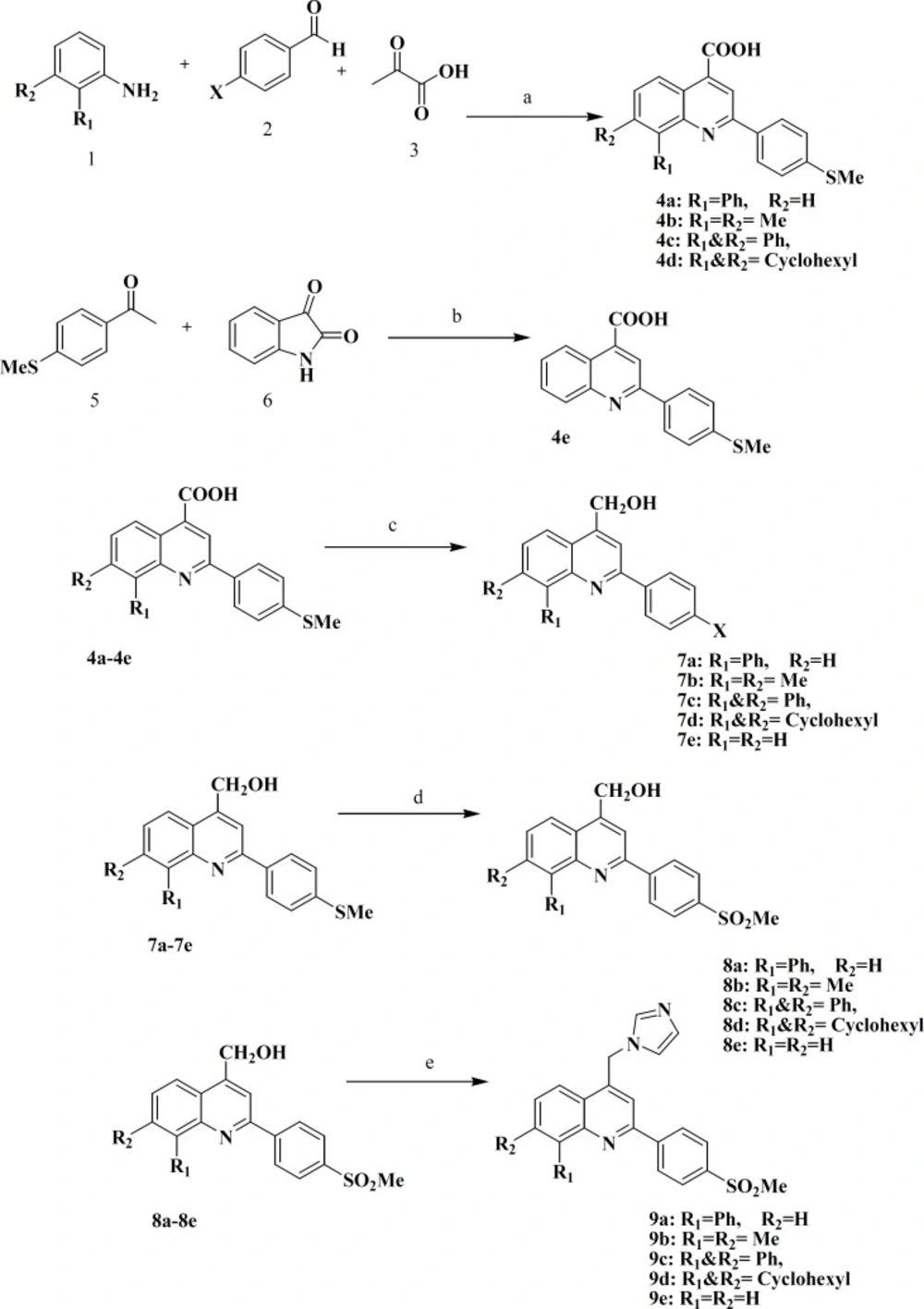
Chemistry
All chemicals and solvents used in this study were purchased from Merck AG and Aldrich Chemical. Melting points were determined with a Thomas-Hoover capillary apparatus. Infrared spectra were acquired using a Perkin Elmer Model 1420 spectrometer. A Bruker FT-500 MHz instrument (Brucker Biosciences, USA) was used to acquire 1H NMR spectra with TMS as internal standard. Chloroform-D and DMSO-d6 were used as solvents. Coupling constant (J) values are estimated in hertz (Hz) and spin multiples are given as s (singlet), d (double), t (triplet), q (quartet), m (multiplet), and br (broad). The mass spectral measurements were performed on an 6410Agilent LCMS triple quadrupole mass spectrometer(LCMS) with an electrospray ionization (ESI) interface. Microanalyses, determined for C and H, were within ± 0.4% of theoretical values.
General procedure for preparation of 2-(4-(methylthio)phenyl)-7,8-substituted-quinoline-4-carboxylic acid (4a-4e)
These compounds were synthesized according to our pervious methods(
7,
11).
General procedure for preparation of 2-(4-methylthio-phenyl)-7,8-substituted-quinoline-4-yl)methanol (7a-7e)
LiAlH
4 (0.45 g, 12 mmol) was suspended in dry THF (20 ml) under a nitrogen atmosphere. Under vigorous stirring a solution of appropriate acid(4a-4e) (5.67 mmol) in dry THF was added dropwise to keep the reaction mixture slightly boiling. After stirring for 2 h at room temperature, the suspension was carefully hydrolyzed with NaOH solution (10%) till no more hydrogen was produced. The solid was filtered off and washed thoroughly with chloroform(
12). The filtrate was dried over Na
2SO
4 and the solvent was removed under reduced pressure to yield a yellow oil which was crystallized from ether/hexane (25:75 v/v) (yield: 62-79 %).
2-(4-Methylthio-phenyl)-8-phenylquinoline-4-yl)methanol (7a)
Yield: 77%; yellow crystalline powder; mp=131-133˚C; IR (KBr): ν (cm-1), 3745-2371(OH);LCMS (ESI): 380.6 (M+23) +100.
2-(4-Methylthio-phenyl)-7,8-dimethylquinoline-4-yl)methanol (7b)
Yield: 62%; pale yellow crystalline powder; mp=140-141˚C; IR (KBr): ν (cm-1), 3406(OH);
LCMS (ESI): 310.2 (M+1) +100.
2-(4-Methylthio-phenyl)-benzo[h]quinoline-4-yl)methanol (7c)
Yield: 68%; yellow crystalline powder; mp=111-113˚C; IR (KBr): ν (cm-1), 3290(OH);
LCMS (ESI): 332.9 (M+1) +100.
7,8,9,10-Tetrahydro-2-(4-methylthio-phenyl)-benzo[h]quinoline-4-yl)methanol (7d)
Yield: 79%; pale yellow crystalline powder; mp=139-140ºC; IR (KBr): ν (cm-1), 3234(OH);
LCMS (ESI): 336.9 (M+1) +100.
2-(4-Methylthio)phenyl- quinoline-4-yl)methanol (7e)
Yield: 63%; yellow crystalline powder; mp=120-122ºC; IR (KBr): ν (cm-1), 3224(OH);
LCMS (ESI): 282.1 (M+1) +100.
General procedure for preparation of 2-(4-methylsulfonyl-phenyl)-7,8-substituted-quinoline-4-yl)methanol (8a-8e)
One gram of 2-(4-methylthio)phenyl-7,8-substituted-quinoline-4-yl)methanol (7a-7e)was dissolved in 10 ml THF and 5 g oxone in THF/water was added. The mixture was stirred at room temperature for 3-5 h, after evaporation of THF, the residue was extracted with chloroform and dried with sodium sulfate and then evaporated(
13), the product was recrystallized in chloroform/hexane (yields: 67-70%).
2-(4-Methylsulfonyl-phenyl)-8-phenylquinoline-4-yl)methanol (8a)
Yield: 48%; yellow crystalline powder; mp=178-179ºC; IR (KBr): ν (cm-1), 3442(OH) 1312, 1157(SO2);LCMS (ESI): 390.8 (M+1) +100.
2-(4-Methylsulfonyl-phenyl)-7,8-dimethylquinoline-4-yl)methanol (8b)
Yield42%; orange crystalline powder; mp=148-150ºC; IR (KBr): ν (cm-1), 3241(OH), 1303, 1149(SO2);LCMS (ESI): 342.1(M+1) +100.
2-(4-Methylsulfonyl-phenyl)-benzo[h]quinoline-4-yl)methanol (8c)
Yield: 65%; yellow crystalline powder; mp=210-211ºC; IR (KBr): ν (cm-1), 3485 (OH), 1296, 1145(SO2);1HNMR (DMSO-d6 ): δ(ppm) 3.28(s, 3H, SO2Me), 5.18 (s, 2H, CH2), 5.71 (s, 1H, OH), 7.74-7.80 (m, 2H, benzoquinoline H8&H9), 7.96-7.80(m, 2H,benzoquinoline H6&H5), 7.99 (d, 1H,benzoquinoline H7) 8.11 (d, 2H, 4-methylsulfonylphenyl H2&H6, J=8.46Hz), 8.37 (s, 1H,benzoquinoline H3), 8.63 (d, 2H, 4-methylsulfonylphenyl H3&H5, J=8.46Hz), 9.4 (d, 1H, quinoline H10, J=7.34Hz); LCMS(ESI): 364.1 (M+1)+100.
7,8,9,10-Tetrahydro-2-(4-methylsulfonyl-phenyl)-benzo[h]quinoline-4-yl)methanol (8d)
Yield: 57%; yellow crystalline powder; mp=162-163ºC; IR (KBr): ν (cm-1), 3365 (OH), 1309, 1159(SO2);LCMS (ESI): 368.9 (M+1) +100.
2-(4-Methylsulfonyl)phenyl-quinoline-4-yl)methanol (8e)
Yield: 75%; yellow crystalline powder; mp=164-166ºC IR (KBr): ν (cm-1), 3340(OH), 1301, 1151(SO2);LCMS (ESI): 314.9 (M+1) +100.
General procedure for preparation of 4-((1H-imidazol-1-yl)methyl)-2-(4-methylsulfonyl-phenyl)-7,8-substituted-quinoline
To a solution of the corresponding alcohol (1.59 mmol) in 15 ml NMP (N-methyl-2-pyrrolidone), CDI (carbonyl 1, 1-diimidazole) (1.29 g, 7.9 mmol) was added. Then the solution was heated to reflux for 20 h at 170 ºC. After cooling to ambient temperature, it was diluted with water (50 ml) and extracted with ethyl acetate. The combined organic phases was washed with brine and water, dried over Na
2SO
4, and evaporated under reduced pressure (
14). Then the desired product was purified by flash chromatography on silica gel (dichloromethane/methanol, 90:10 v/v), (yield: 13-62%).
4-((1H-Imidazol-1-yl)methyl)-2-(4-methylsulfonyl phenyl)-8-phenyl-quinoline (9a)
Yield: 25%; cream crystalline powder; mp=196-198ºC;IR (KBr): ν (cm-1), 1310, 1150 (SO2);1HNMR (CDCl3 ):δ(ppm) 3.03 (s, 3H, SO2Me) 5.73 (s, 2H, CH2), 7.04 (s, 1H, imidazole H5), 7.23(s, 1H, imidazole H2), 7.34(s, 1H, imidazole H4), 7.45 (t, 1H, phenyl H4, J=7.15Hz), 7.52 (t, 2H, phenyl H3&H5, J=7.31Hz), 7.69-7.76(m, 4H, quinoline H6&H7&phenyl H2 &H6), 7.86 (d, 1H, quinoline H5, J=6.88 Hz ),7.92 (s, 1H, quinoline H3), 7.95 (d, 2H, 4-methoxysulfonylphenyl H2&H6, J=8.13Hz), 8.14 (d, 2H, 4-methoxysulfonylphenyl H3&H5, J=8.13Hz); LCMS(ESI): 440.6 (M+1)+100.
4-((1H-Imidazol-1-yl)methyl)-2-(4- methylsulfonyl phenyl)-7,8-dimethyl-quinoline (9b)
Yield: 61%; yellow crystalline powder; mp=218-219ºC; IR (KBr): ν (cm-1), 1301, 1150 (SO2); 1HNMR (CDCl3 ): δ(ppm) 2.58(s, 3H, Me), 2.91(s, 3H, Me), 3.12(s, 3H, SO2Me), 5.76 (s, 2H, CH2), 7.05 (s, 1H, imidazole H5), 7.26(s, 1H, imidazole H4), 7.39(s, 1H, imidazole H2), 7.50 (d, 1H, quinoline H6, J=8.53Hz), 7.73 (d, 1H, quinoline H5,J=8.53Hz), 8.01 (s, 1H, quinoline H3), 8.08 (d, 2H, 4-methylsulfonylphenyl H2&H6, J=8.53Hz), 8.26 (d, 2H, 4-methylsulfonylphenyl H3&H5, J=8.53Hz); LCMS(ESI): 392.1 (M+1)+100.
4-((1H-Imidazol-1-yl)methyl)-2-(4- methylsulfonyl phenyl)-benzo[h]-quinoline (9c)
Yield: 25%; cream crystalline powder; mp=216-217ºC; IR (KBr): ν (cm-1) 1303, 1148 (SO2);1HNMR (DMSO-d6 ): δ(ppm) 3.26(s, 3H, SO2Me), 5.90 (s, 2H, CH2 ), 7.01 (s, 1H, imidazole H5), 7.36(s, 1H, imidazole H2), 7.79 (m, 2H, benzoquinoline H8&H9), 7.93(s, 1H, imidazole H4), 8.03-8.05(m, 3H,benzoquinoline H3&H6&H7), 8.10 (d, 2H, 4-methylsulfonylphenyl H2&H6, J=8.32Hz), 8.14 (d, 1H,benzoquinoline H10), 8.50 (d, 2H, 4-methylsulfonylphenyl H3&H5, J=8.32Hz), 9.4 (d, 1H, quinoline H5); LCMS(ESI): 414.6 (M+1)+100.
4-((1H-Imidazol-1-yl)methyl)-7,8,9,10-tetrahydro-2-(4-methylsulfonylphenyl)-benzo[h]-quinoline (9d)
Yield: 35%; cream crystalline powder; mp=247-248ºC; IR (KBr): ν (cm-1) 1318, 1163(SO2);1HNMR (CDCl3 ): δ(ppm) 1.9-2.01(m, 4H, CH2), 2.96 (m, 2H, CH2), 3.08 (s, 3H, SO2Me), 3.46 (m, 2H, CH2),5.67 (s, 2H, CH2 ), 7.0 (s, 1H, imidazole H5 ), 7.2(s, 1H, imidazole H2 ), 7.29 (s, 1H, imidazole H4 ), 7.35 (d, 1H, quinoline H6 J=8.57 Hz), 7.66 (d, 1H, quinoline H5, J=8.58 Hz), 7.70 (s, 1H, quinoline H3), 8.03 (d, 2H, 4-methoxysulfonylphenyl H2&H6, J=8.46Hz), 8.29 (d, 2H, 4-methoxysulfonylphenyl H3&H5, J=8.46Hz); LCMS(ESI): 418.7 (M+1)+100.
4-((1H-Imidazol-1-yl)methyl)-2-(4- methylsulfonyl phenyl)-quinoline (9e)
Yield: 15%; cream crystalline powder; mp=189-190ºC; IR (KBr): ν (cm-1) 1300, 1150(SO2);1HNMR (CDCl3 ): δ (ppm) 3.1(s, 3H, SO2Me), 5.76 (s, 2H, CH2), 7.07 (s, 1H, imidazole H5), 7.26(s, 1H, imidazole H4), 7.29(s, 1H, imidazole H2), 7.69-7.72 (m, 2H, quinoline H6&H3), 7.87 (t, 1H, quinoline H7, J=7.27Hz), 7.98 (d, 1H, quinoline H5, J=8.28Hz), 8.09 (d, 2H, 4-methylsulfonylphenyl H2&H6, J=8.37Hz), 8.26 (d, 2H, 4-methylsulfonylphenyl H3&H5, J=8.37Hz), 8.3 (d, 1H, quinoline H8, J=8.40Hz); LCMS(ESI): 364.8 (M+1)+100.
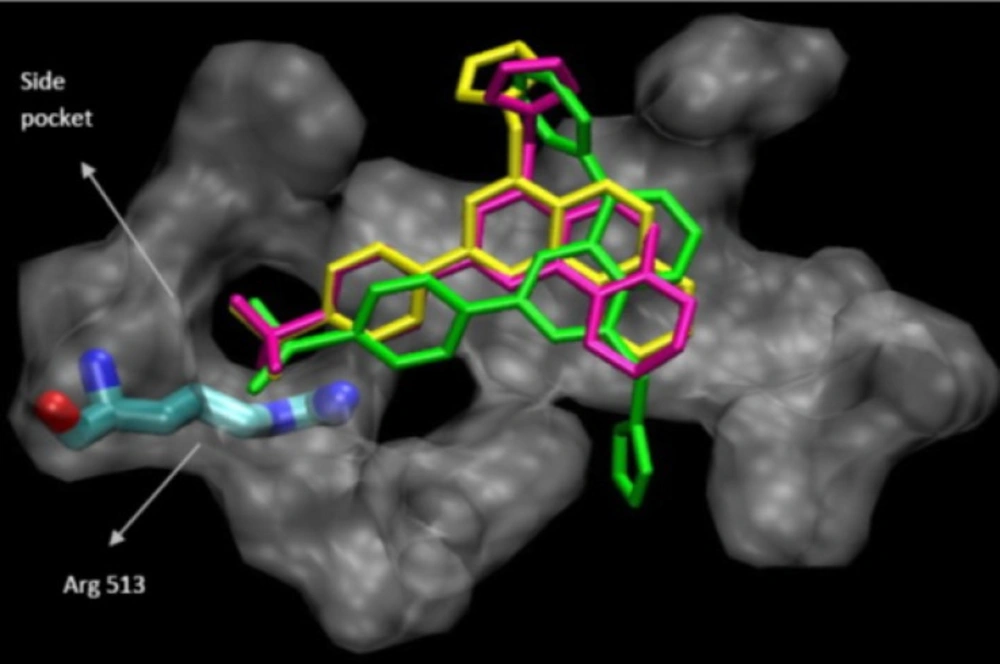
Molecular modeling (docking) studies
Docking studies were performed using Autodock software Version 3.0. The coordinates of the X-ray crystal structure of the selective COX-2 inhibitor SC-558 bound to the murine COX-2 enzyme was obtained from the RCSB Protein Data Bank (1cx2) and hydrogens were added. The ligand molecules were constructed using the Builder module and were energy minimized for 1000 iterations reaching a convergence of 0.01 kcal/mol.The energy minimized ligands were superimposed on SC-558 in the PDB file 1cx2 after which SC-558 was deleted. The purpose of docking is to search for favorable binding configuration between the small flexible ligands and the rigid protein. Protein residues with atoms greater than 7.5 A from the docking box were removed for efficiency. These docked structures were very similar to the minimized structures obtained initially. The quality of the docked structures was evaluated by measuring the intermolecular energy of the ligand–enzyme assembly (
15,
16).
In-vitro cyclooxygenase (COX) inhibition assays
The assay was performed using an enzyme chemiluminescentkit (Cayman Chemical, MI, USA) according to our previously reportedmethod(
17)The Cayman chemical chemiluminescent COX
(ovine) inhibitor screening assay utilizes the heme-catalyzedhydroperoxidase activity of ovine cyclooxygenases to generateluminescence in the presence of a cyclic naphthalene hydrazideand the substrate arachidonic acid. Arachidonate-induced luminescencewas shown to be an index of real-time catalytic activity anddemonstrated the turnover inactivation of the enzyme. Inhibition
of COX activity, measured by luminescence, by a variety of selectiveand non-selective nhibitors showed potencies similar to those observedwith other in-vitro and whole cell methods.
Cytotoxicity assay
Cell line and culture conditions
The human breast cancer T47D and MCF-7 cell lines were obtained from Pasteur Institute Cell Bank of IRAN (Tehran, IRAN). Cells were maintained in RPMI-1640(Gibco, UK) culture medium supplemented with 10% fetal bovine serum (Gibco, UK) and 100 U Ml-1 of penicillin and 100 ng Ml-1 of streptomycin (Gibco, UK) at 37 C in 5% CO2 incubator. All reagent and chemicals used in this experiment were of cell culture or molecular biology grade, purchased from different international sources.
General procedure
The MTT (3-[4, 5-dimethylthiazol-2-yl]-2,5-diphenyl tetrazolium bromide) based assay was performed by seeding 5000 cells (T47D and MCF-7) per 180 µL RPMI complete culture medium in each well of 96-well culture plates. The day after seeding, culture medium was changed with medium containing standard anti-tumor drug Doxorubicin as well as different concentrations of newly synthesized compounds and RPMI control (no drug). Cells were then incubated at 37ºC in 5% Co2 incubator for 48h and 72h. Then 25 µL of MTT solution (4mg Ml
-1) were added to each well and further incubated at 37ºC for 3h. At the end of incubation, formazan crystals were dissolved in 100 µL of DMSO and plates were read in a plate reader (TECAN, Austria) at 540 nm. This experiment was performed in triplicate determination each time (
18,
19).