Method development and optimization
The electrochemical behaviour of fulvestrant was investigated at the Pt disc electrode in acetonitrile solution containing 0.1 M LiClO
4 as the supporting electrolyte by using cyclic voltammetry (CV).
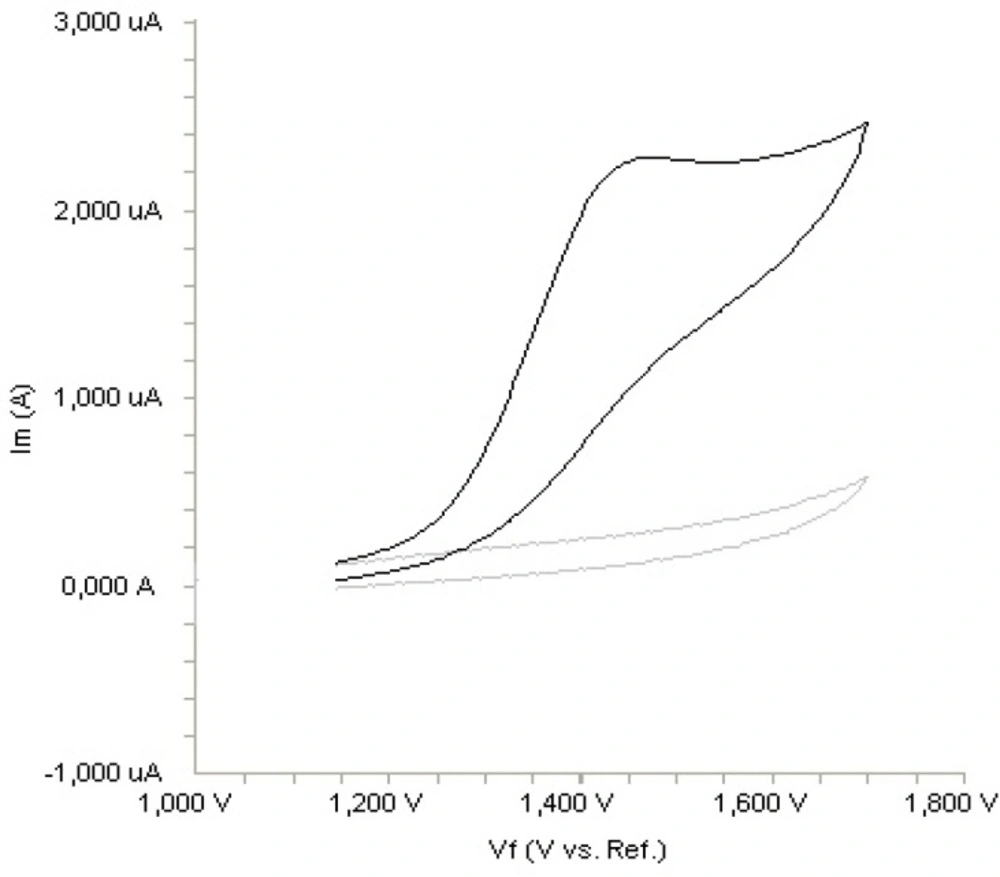
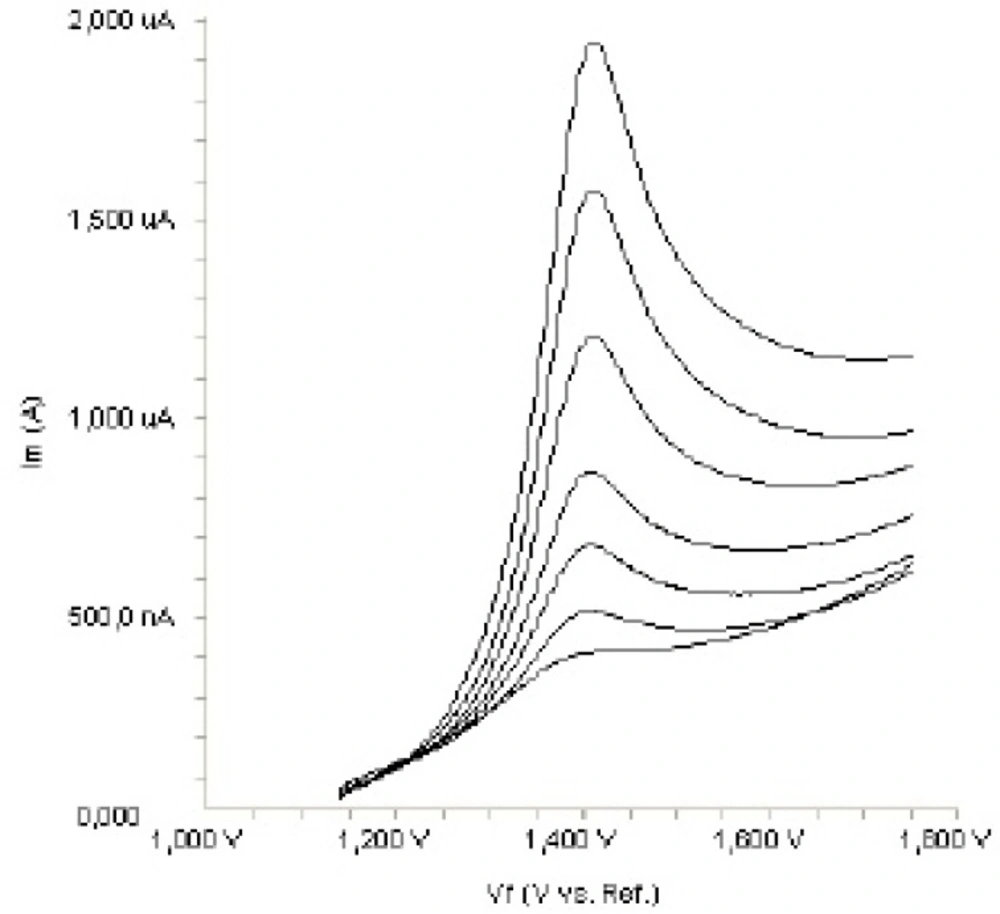
Figure 2. shows a typical cyclic voltammogram of 20 m g mL
-1 fulvestrant recorded under these conditions for the scan rate of 0.1 V s
-1. In the anodic sweep, an oxidation peak is seen at about potential of 1.4 V. Upon reversing the potential scan, no reduction peak corresponding to this oxidation wave is observed, indicating the irreversible nature of the electrode reactions.
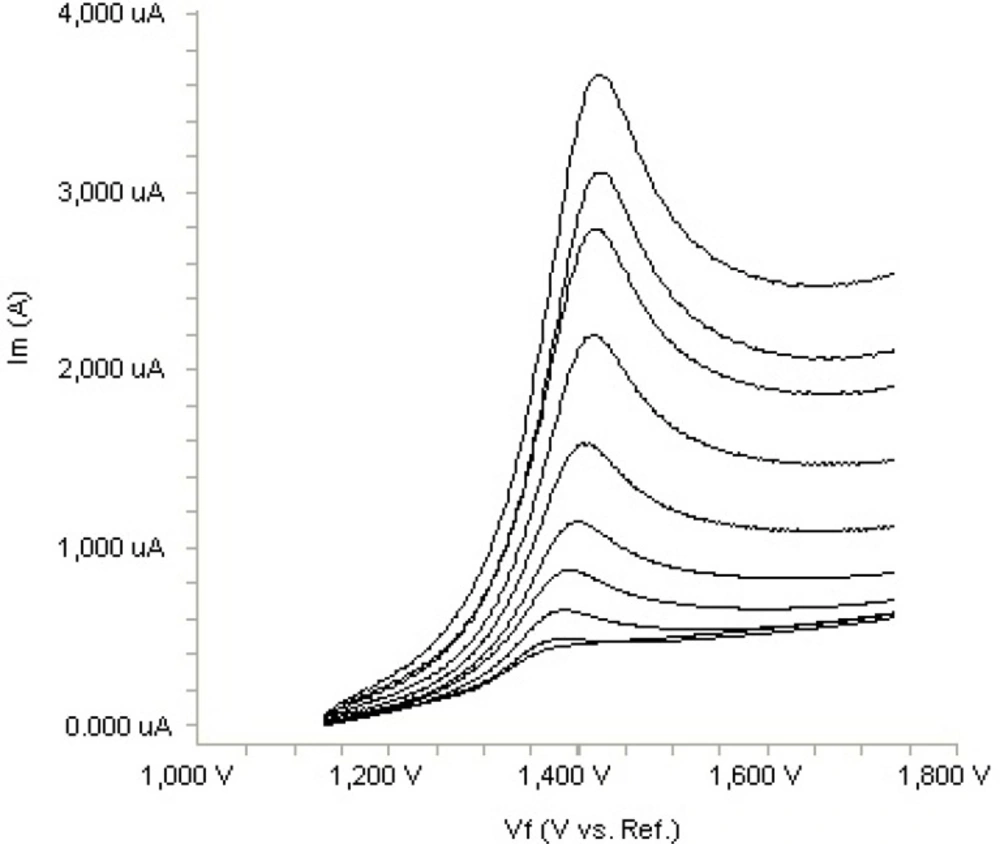
In order to gain a deeper insight into the voltammetric waves, the effect of scan rate on the anodic peak currents (I
m) and peak potentials (E
p) was studied in the range of 0.01-1 V s
-1 of the potential scan rates in acetonitrile solution containing 20 m g mL
-1 concentration of fulvestrant ( 3.). The representative linear sweep voltammograms obtained at Pt electrode for 20 m g mL
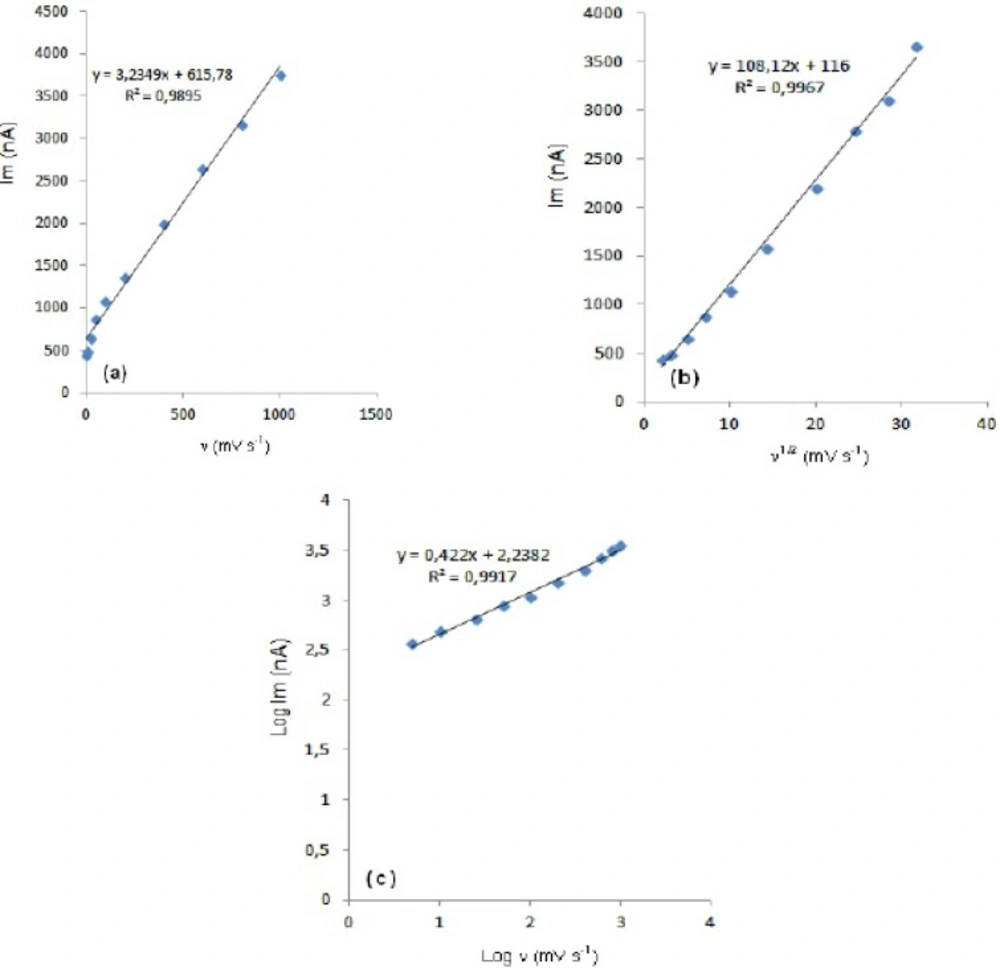
-1 concentration of fulvestrant display straight lines with 0.42 slope (
Figure 4c), which are close to theoretical value of 0.5 expected for an ideal diffusion-controlled electrode process.
14 log I
m-log ν curve is more eligible for this aim, therefore, a diffusional process for peak should be considered. These results suggest that the redox species are diffusing freely from the solution and not precipitating onto the electrode surface. The reason for this behaviour may be due to the solubility of the intermediate species in acetonitrile or poor adherence of products on the electrode surface.As shown in
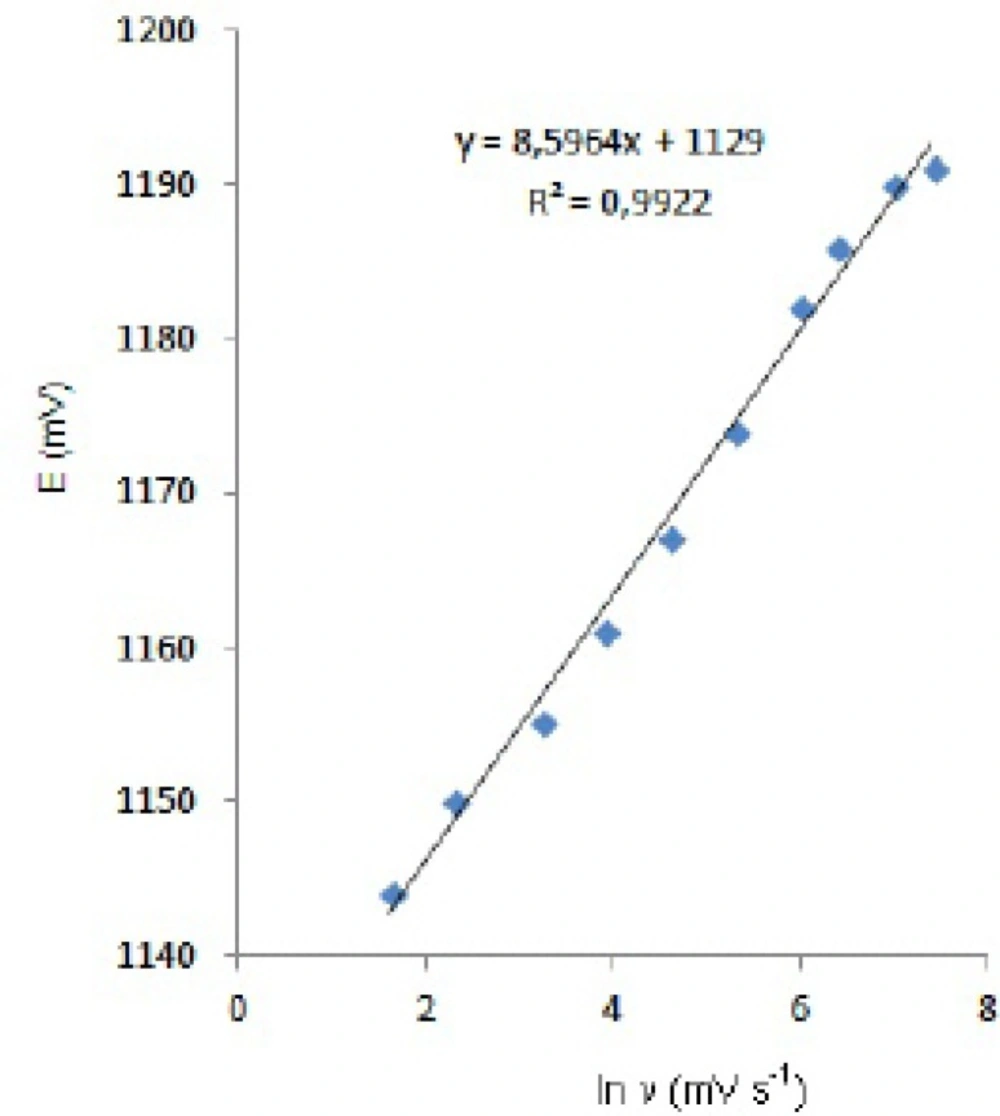
Figure 3. the oxidation peak potential (E
pa) for peaks shift toward more positive values with increasing scan rate. The relationship between the peak potential and scan rate is described by the following equation,
and from the variation of peak potential with scan rate αn
a can be determined, where α is the transfer coefficient and n
a is the number of electrons transferred in the rate determining step. According to this equation, the plots of the peak potentials versus ln ν for oxidation peak show linear relationship (
Figure 5). The slope indicates the value of αn
a is 0.75 for peak. Also, this value obtained indicates the total irreversibility of the electron transfer processes. This result shows that the chemical step is a fast following reaction coupled to a charge transfer
During HPLC method development, different organic solvents were tested as mobile phase. The best peak was achieved with the mixture of 1% orthophosphoric acid-methanol (80:20, v/v). Chromatographic separation was achieved with reverse phase C18 analytical column. When the different wavelength was investigated with UV detection, the best chromatogram for fulvestrant was obtained at 243 nm wavelength.
Method validation
To ensure the optimization of the methods in light of the standardization rules, we developed these methods along with the process of validation. The assay methods were evaluated through the determination of specificity, linearity, precision, accuracy, limit of detection, limit of quantification, recovery and the stability effect was investigated by analysing the pure fulvestrant solution and drug sample (
15).
Specificity
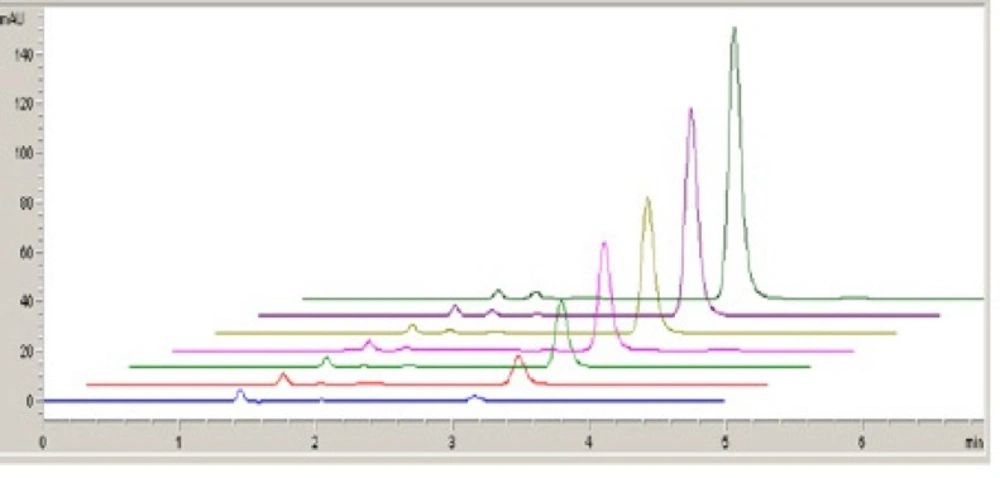
All the solutions were scanned from 1.0 to 1.7 V and checked for change in the peaks at respective potentials (
Figure 6).
In a separate study, the specificity of the method was investigated by observing interferences between the fulvestrant and excipients. The retention time of fulvestrant in HPLC method was approximately 3.1 min with good peak shape (
Figure 7).
Linearity
For LSV and HPLC measurements, the solutions were prepared by dilution of the stock solution of fulvestrant to reach a concentration range of 5-50 m g mL
-1 (5, 10, 15, 20, 30, 40 and 50 m g mL
-1) and 0.5-20 m g mL
-1 (0.5, 1, 2, 5, 10, 15 and 20 m g mL
-1), respectively. Calibration curves were constructed for fulvestrant standard by plotting the concentration of fulvestrant versus voltammogram and peak area response. The calibration curve constructed was evaluated by its correlation coefficient. The correlation coefficient (
r) of all the calibration curves were consistently greater than 0.99. The regression equations were calculated from the calibration graphs, along with the standard deviations of the slope and intercept on the ordinate. The results are shown in
Table 1.
Precision and accuracy
The precision of the LSV and HPLC methods was determined by repeatability (intra-day) and intermediate precision (inter-day). Repeatability was evaluated by analysing QC samples six times per day, at three different concentrations which were QC samples. The intermediate precision was evaluated by analysing the same samples once daily for two days. The RSD of the predicted concentrations from the regression equation was taken as precision (16-19). The accuracy of this analytic method was assessed as the percentage relative error. For all the concentrations studied, intra- and inter-day relative standard deviation values were £ 2.66%. These results were given in
Table 2.
Limits of detection (LOD) and quantification (LOQ)
For LSV measurements, LOD and LOQ of the fulvestrant were determined using calibration standards. The LOD and LOQ values were calculated as 3.3
σ/
S and 10
σ/
S, respectively, where
S is the slope of the calibration curve and
σ is the standard deviation of
y-intercept of regression equation (
n = 6) (
20).
For HPLC measurements, the LOD and LOQ of the fulvestrant were determined by injecting progressively low concentration of the standard solution under the chromatographic conditions. The lowest concentrations assayed where the signal/noise ratio was at least 10:1, this concentration was regarded as LOQ. The LOD was defined as a signal/noise ratio of 3:1. The LOD and LOQ for LSV were 1.52 and 5.0 m g mL-1, for HPLC 0.152 and 0.50 m g mL-1, respectively. Among the two methods, HPLC is more sensitive than LSV.
Recovery
To determine the accuracy of the LSV and HPLC methods and to study the interference of formulation additives, the recovery was checked as three different concentration levels. Analytical recovery experiments were performed by adding the known amount of pure drugs to pre-analyzed samples of commercial dosage form. The recovery values were calculated by comparing the concentration obtained from the spiked samples with actual added concentrations. These values are also listed in
Table 3.
Ruggedness
In this study, the LSV and HPLC determination of fulvestrant were carried out by a different analyst in the same instrument with the same standard (
Table 4). The results showed no statistical differences between different operators suggesting that the developed method was rugged.
Stability
Stability studies indicated that the samples were stable when kept at room temperature, +4 0 C and -20 0 C refrigeration temperature for 24 h (short-term) and refrigerated at +4 and -20 0 C for 72 h (long-term). There was no significant change in the analysis over a period of 72 h. The mean RSD between peak areas for the samples stored under refrigeration (4 ± 1 ° C), at room temperature (25 ± 1 ° C) and refrigeration (-20 ± 1 ° C) were found to be 1.47%, 1.78% and 1.92%, respectively, suggesting that the drug solution can be stored without any degradation over the studied time interval.
Also, The ICH guideline entitled stability testing of drug substances and products requires the stress testing to be carried out to elucidate the inherent stability characteristics of the active substance, and provide a rapid identification of differences that might result from changes in the manufacturing processes or source sample (
19). Susceptibilities to acid, alkali and oxidation hydrolysis stability are the required tests.
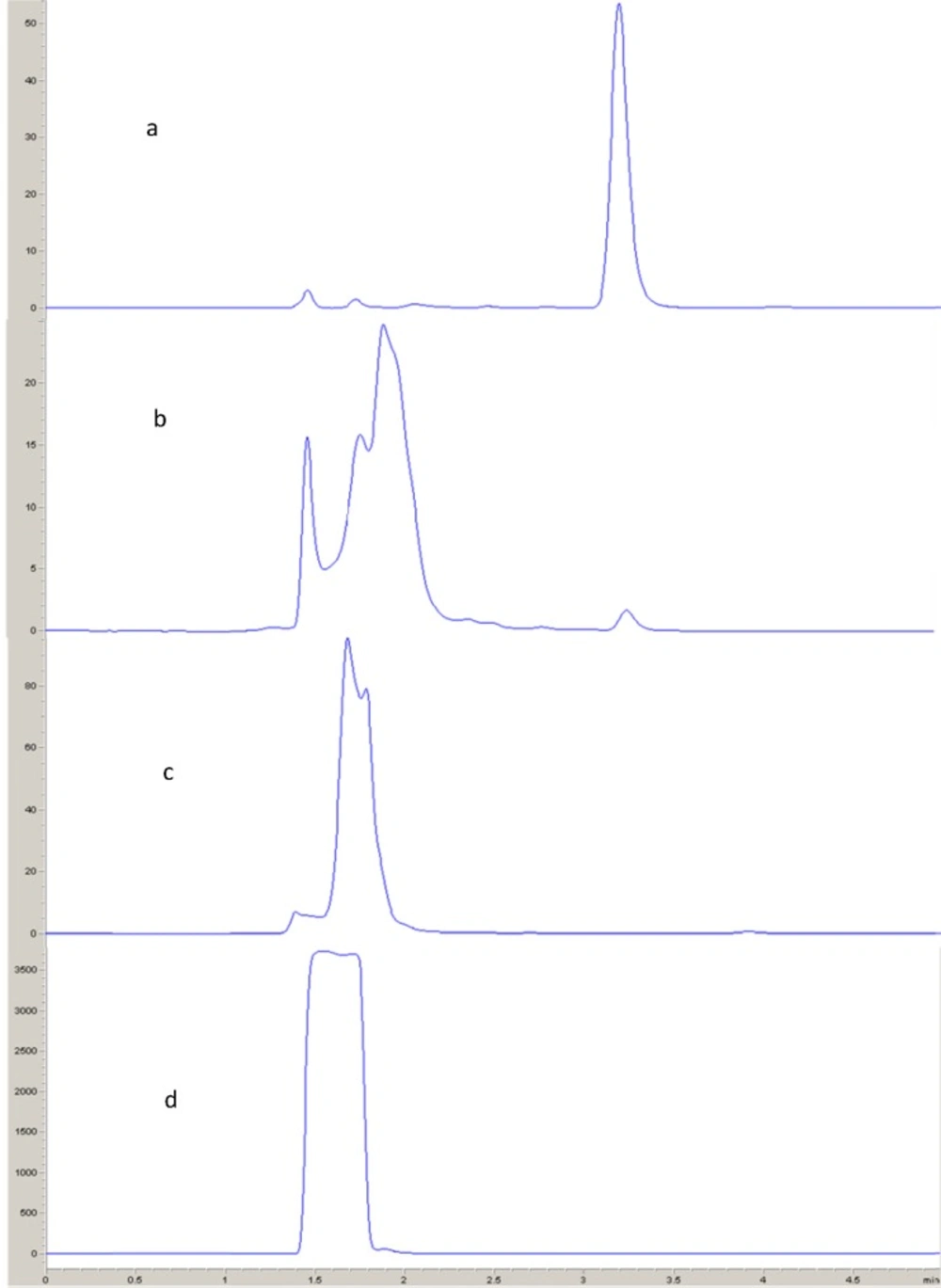
Acid and alkali hydrolysis
Aliquot of 0.2 mL of fulvestrant solution (50 m g mL
-1) was transferred to a small rounded flask. The solution was mixed with 0.8 mL of 0.1 N hydrochloric acid, or 0.1 N sodium hydroxide. The prepared solutions were subjected to reflux for 2 h in a boiling water bath. The samples were cooled to room temperature (25 ± 5 ° C), neutralized with an amount of acid or base equivalent to that of the previously added. From the resulting neutral solution, 10 μL was injected into the HPLC system (
Figure 8).
Oxidation
0.2 mL of fulvestrant solution (50 m g mL
-1) was transferred to rounded flask. The contents were then mixed with 0.8 mL of 30% hydrogen peroxide solution, and the reaction mixture was allowed to proceed at room temperature (25 ± 5 ° C) for 2 h with intermittent shaking. A volume of 10 μL was injected into the HPLC system (
Figure 8).
Comparison of the methods
The first paper related to electrochemical investigation of the fulvestrant has been reported by Dogan Topal and Ozkan (
21). In this paper, the electrochemical oxidation of the fulvestrant has been studied by means of cyclic voltammetry and differential pulse voltammetry in pH 4.80 acetate buffer (30% ethanol) at a modified pencil graphite electrode, and it was reported that the drug exhibited an irreversible and diffusion-controlled oxidation peak. According to the molecular structure, literature knowledge and the obtained results, the oxidation mechanism of the fulvestrant may be postulated by an initial oxidation with two electrons and the conversion of hydroxyl group to quinone, which was electro-active in both acidic and basic media (
22,
23). Also, LSV and HPLC methods were applied for the determination of fulvestrant in Faslodex injectable solution. The results show the high reliability and reproducibility of two methods. The results were statistically compared using the F-test. At 95% confidence level, the calculated
F-values do not exceed the theoretical values (
Table 5) Therefore, there is no significant difference between LSV and HPLC method.
Also, the suggested LSV and HPLC methods were compared with the reported differential pulse voltammetry (
21). There was no significant difference between the three methods with respect to mean values and standard deviations at the 95% confidence level (
Table 5). Therefore, it is suggested that the two methods are equally applicable
.