
Introduction
AKT1/PKBa (protein kinase B) is a serine/threonine kinase that belongs to the AKT family AKT1 (RAC-alpha serine/threonine-protein kinase is an enzyme that in humans is encoded by the AKT1), also known as «AKT» or protein kinase B (PKB) is an important molecule in mammalian cellular signaling. In humans, there are three genes in the «AKT family»: AKT1, AKT2, and AKT3. These enzymes are members of the serine/threonine-specific protein kinase family (1).
AKT1 is activated in cells in response to diverse stimuli such as hormones, growth factors and extracellular matrix components and is involved in glucose metabolism, transcription, survival, cell proliferation, angiogenesis, and cell motility (1, 2). AKT1 is frequently overexpressed and activate in many types of human cancers including cancers of colon, breast, brain, pancreas and prostate as well as lymphomas and leukemias (3, 4).
Docking is an important in the study of protein ligand interaction properties such as binding energy, geometry complementarity, hydrogen bond donor acceptor, hydrophobicity, electron distribution and polarizability thus it plays a major role in the drug discovery for the identification of suitable molecular scaffold and distinguishing selectivity for the target protein (5).
GOLD, the first algorithm to be evaluated on a large data set of complex posse, an empirical free energy scoring function that estimates the free energy of binding permitting inhibition constant for protein ligand complex. It is a package of program for structure visualization and manipulation for docking, the post processing and visualization of the results (6). The objective of the present work is to study the in silico of AKT1inhibitory activity of some new synthetic N1, N4-bis ((2-chloroquinolin-3-yl) methylene) benzene-1, 4-diamine.
Quinoline and its derivatives have always attracted both synthetic and biological chemists because of its diverse chemical and pharmacological properties (7). For example quinine has been used for the treatment of malaria (8), dynemicin A and streptonigrin, naturally occurring members of the class of antitumor antibiotic (9, 10). According to our literature review we find that compounds containing quinoline (11-14), have been reported to exhibit antiinflammatory activity (15). To study the combined effect of these two moieties (quinoline and diimine) in a single network, there is an interest in the synthesis of N1, N4-bis ((2-chloroquinolin-3-yl) methylene) benzene-1, 4-diamine (3a-3i). Therefore, we studied our compound as a potential inhibitor for AKT1 enzyme.
Experimental
Materials and Methods
The products were characterized by spectroscopic data (IR, 1H NMR, elemental analyses). The purity determinations of the products were accomplished by TLC on silica gel polygram STL G/UV 254 plates. Melting points were determined with an Electrothermal Type 9100 melting point apparatus. Elemental analyses were made by a Thermo Finning Flash EA1112 CHNO-S analyzer and agreed with the calculated values. The FTIR spectra were recorded on an Avatar 370 FTIR Therma Nicolet spectrometer. The NMR spectra were recorded on a Bruker Avance 100 and 400 MHz instrument in CDCl3 and DMSO.
General experimental procedure for the preparation of N1, N4-bis ((2-chloroquinolin-3-yl) methylene) benzene-1, 4-diamine (3a-i).
A mixture of 2-chloroquinoline-3-carbaldehydes (3 mmol, 0.5734g) (1), benzene-1,4-diamine (1.5 mmol, 0.1622 g) (2), few drops glacial acetic acid and 40 mL EtOH in a 100 mL flask was stirred at reflux for 4 h. After completion of the reaction (monitored by TLC, ethyl acetate/n-hexane, 1/1), the resulting solid was separated by filtration, and recrystallized from ethanol to afford pure product.
N1, N4-Bis ((2-chloroquinolin-3-yl) methylene) benzene-1, 4-diamine (3a)
Yield: 72%; mp 195 °C; IR (KBr) υcm-1: 3000-3300 (CH aromatic), 2952 (CH aliphatic), 1616 (C=N imine), 1557 (C=N quinoline), 1520 (C=C quinoline), 1500 (C=C phenyl), 1057 (C-Cl quinolone ring). 1H NMR (400 MHz, CDCl3, 25 °C, ppm) δ: 7.46 (s, 4H, meddle benzene ring), 7.65 (m, 2H, H7), 7.84 (m, 2H, H6), 8.09 (d, 2H, J=8.4, H5), 8.10 (dd, 2H, J=8.8, 0.8, H8), 9.09(s, 2H, HC=N), 9.10(s, 2H, H4). Anal. Calcd. for C26H16Cl2N4: C, 68.58; H, 3.54; Cl,15.57; N, 12.30; Found: C, 68.54; H, 3.57; N, 12.28.
N1, N4-Bis ((2-chloro-6-methylquinolin-3-yl) methylene) benzene-1, 4-diamine (3b)
Yield: 70%; mp 212-215 °C; IR (KBr) υ cm-1: 3100 (CH aromatic), 2900-3000 (CH aliphatic), 1619 (C=N imine), 1580 (C=N quinoline), 1525 (C=C quinoline), 1504 (C=C phenyl), 1059 (C-Cl quinolone ring). 1H NMR (400 MHz, DMSO, 25 °C, ppm) δ: 2.46 (s, 6H, CH3), 6.36 (s, 4H, meddle benzene ring), 7.24 (d, 2H, J=8.75, H7), 7.86 (d, 2H, J=8.75, H8), 7.88 (s, 2H, H5), 7.95 (s, 2H, HC=N), 8.87 (s, 2H, H4). Anal. Calcd. for C28H20Cl2N4: C, 69.57; H, 4.17; Cl,14.67; N, 11.59; Found: C, 69.59; H, 4.20; N, 11.62.
N1, N4-Bis ((2-chloro-6-methoxyquinolin-3-yl) methylene) benzene-1, 4-diamine (3c)
Yield: 69%; mp 218-220 °C; IR (KBr) υ cm-1: 3100-3200 (CH aromatic), 2900-3000 (CH aliphatic), 1615 (C=N imine), 1572 (C=N quinoline), 1518 (C=C quinoline), 1498(C=C phenyl), 1234(C-O), 1055(C-Cl quinolone ring). 1H NMR (400 MHz, DMSO, 25 °C, ppm) δ: 3.01 (s, 6H, CH3), 6.37 (s, 4H, meddle benzene ring), 6.61 (d, 2H, J=8.75, H7), 7.22 (d, 2H, J=8.75, H8), 7.62 (s, 2H, H5), 7.88 (s, 2H, HC=N), 8.87 (s, 2H, H4). Anal. Calcd. for C28H20Cl2N4O2: C, 65.25; H, 3.91; Cl,13.76; N, 10.87; Found: C, 65.21; H, 3.90; N, 10.89.
N1, N4-Bis ((2-chloro-6-ethoxyquinolin-3-yl) methylene) benzene-1, 4-diamine (3d)
Yield: 74%; mp 196 °C; IR (KBr) υ cm-1: 3100-3150 (CH aromatic), 2895-2950 (CH aliphatic), 1600 (C=N imine), 1570 (C=N quinoline), 1520 (C=C quinoline), 1500 (C=C phenyl), 1234 (C-O), 1057 (C-Cl quinolone ring). 1H NMR (400 MHz, DMSO, 25 °C, ppm) δ: 1.38 (t, 6H, J=6.75, CH3 ), 2.74 (q, 4H, J=6.75, CH2), 6.39 (s, 4H, meddle benzene ring), 6.61 (d, 2H, J=8.5, H7), 7.21 (d, 2H, J=8.5, H8), 7.45 (s, 2H, H5), 7.60 (s, 2H, HC=N), 8.88 (s, 2H, H4). Anal. Calcd. for C30H24Cl2N4O2: C, 66.30; H, 4.45; Cl,13.05; N, 10.31; Found: C, 66.29; H, 4.41; N, 10.28.
N1, N4-Bis ((2, 6 -dichloroquinolin-3-yl) methylene) benzene-1, 4-diamine (3e)
Yield: 74%; mp 196-198 °C; IR (KBr) υ cm-1: 2900-3300 (CH aromatic), 2862 (CH aliphatic), 1620 (C=N imine), 1573 (C=N quinoline), 1519 (C=C quinoline), 1500 (C=C phenyl), 1055(C-Cl quinolone ring). 1H NMR (400 MHz, DMSO, 25 °C, ppm) δ: 6.42 (s, 4H, meddle benzene ring), 6.62 (d, 2H, J=8.75, H7), 7.86 (s, 2H, HC=N), 7.90 (d, 2H, J=8.75, H8), 8.37 (s, 2H, H5), 8.88 (s, 2H, H4). Anal. Calcd. For C26H14Cl4N4: C, 59.57; H, 2.69; Cl, 27.05; N, 10.69; Found: C, 59.60; H, 2.71; N, 10.66.
N1, N4-Bis ((2-chloro-7-methylquinolin-3-yl) methylene) benzene-1, 4-diamine (3f)
Yield: 70%; mp 198 °C; IR (KBr) υ cm-1: 3110-3210 (CH aromatic), 2950 (CH aliphatic), 1612 (C=N imine), 1585 (C=N quinoline), 1519 (C=C quinoline), 1489 (C=C phenyl), 1057 (C-Cl quinolone ring). 1H NMR (400 MHz, CDCl3, 25 °C, ppm) δ: 2.60(s, 6H, CH3), 7.42 (s, 4H, meddle benzene ring), 7.59 (s, 2H, H8), 7.44 (d, 2H. J=8.25, H6), 7.84 (d, 2H, J=8.25, H5), 7.88 (s, 2H, HC=N), 9.02 (s, 2H, H4). Anal. Calcd. for C28H20Cl2N4: C, 69.57; H, 4.17; Cl, 14.67; N, 11.59; Found: C, 69.52; H, 4.12; N, 11.61.
N1, N4-Bis ((2-chloro-6-ethylquinolin-3-yl) methylene) benzene-1, 4-diamine (3g)
Yield: 69%; mp 190 °C; IR (KBr) υ cm-1: 3000-3100 (CH aromatic), 2900-3000 (CH aliphatic), 1610 (C=N imine), 1583 (C=N quinoline), 1517 (C=C quinoline), 1489 (C=C phenyl), 1061 (C-Cl quinolone ring). 1H NMR (400 MHz, CDCl3, 25 °C, ppm) δ: 1.35 (t, 6H, J=7.5, CH3), 2.85 (q, 4H, J=7.5, CH2), 7.42 (s, 4H, meddle benzene ring), 7.67 (d, 2H, J=8.5, H7), 7.96, (d, 2H, J=8.5, H8), 7.73 (s, 2H. H5), 8.42 (s, 2H, HC=N), 9.02 (s, 2H, H4). Anal. Calcd. for C30H24Cl2N4: C, 70.45; H, 4.73; Cl, 13.86; N, 10.59; Found: C, 70.47; H, 4.70; N, 10.94.
N1, N4-Bis ((2-chloro-6-isopropylquinolin-3-yl) methylene) benzene-1, 4-diamine (3h)
Yield: 68%; mp 200 °C; IR (KBr) υ cm-1: 3100-3200 (CH aromatic), 2800-3000 (CH aliphatic), 1615 (C=N imine), 1581 (C=N quinoline), 1515 (C=C quinoline), 1500 (C=C phenyl), 1064 (C-Cl quinolone ring). 1H NMR (400 MHz, CDCl3, 25 °C, ppm) δ: 1.36 (d, J=6.75, 12H, CH3), 3.10 (m, 2H, CH), 7.43 (s, 4H, meddle benzene ring), 7.73 (d, 2H, J=8.75, H7), 7.74 (s, 2H, H5), 7.42 (s, 2H, HC=N), 7.99, (d, 2H, J=8.75, H8), 9.04 (s, 2H, H4). Anal. Calcd. for C32H28Cl2N4: C, 71.24; H, 5.23; Cl, 13.14; N, 10.38; Found: C, 71.24; H, 5.20; N, 10.36.
N1, N4-Bis ((2, 7-dichloroquinolin-3-yl) methylene) benzene-1, 4-diamine (3i)
Yield: 75%; mp 195 °C; IR (KBr) υ cm-1: 3000-3200 (CH aromatic), 2907-3005 (CH aliphatic), 1610 (C=N imine), 1583 (C=N quinoline), 1519 (C=C quinoline), 1477(C=C phenyl), 1060(C-Cl quinolone ring). 1H NMR (400 MHz, DMSO, 25 °C, ppm) δ: 6.48 (s, 4H, meddle benzene ring), 7.70 (d, 2H, J=8.75, H6), 8.24 (d, 2H, J=8.75, H5), 8.04 (s, 2H, H8), 8.85 (s, 2H, HC=N), 8.82 (s, 2H, H4). Anal. Calcd. For C26H14Cl4N4: C, 59.57; H, 2.69; Cl, 27.05; N, 10.69; Found: C, 59.55; H, 2.69; N, 10.72.
Structure optimization
Three dimensional structures of the compounds 3a-i were simulated in Hyper Chem7.5 using MM+ method (RMS gradient = 0.1 kcal mol-1) (HyperChem® Release 7, Hypercube Inc., http://www.hyper.com/). In the second optimization, output files were minimized under Semi empirical AM1 methods (Convergence limit = 0.01; Iteration limit = 50; RMS gradient = 0.1 kcal mol-1; Polak-Ribiere optimizer algorithm) (16, 17).
Crystal structures of AKT1 (EC.2.7.11.1) were retrieved from RCSB Protein Data Bank (PD B entry: 3O96).
Molecular docking
Docking was carried out using GOLD 5.2 (Genetic optimization for Ligand Docking) software based on the Gold Score fitness function, that uses the Genetic algorithm (GA). All water molecules and hetero atoms were omitted from the protein to evaluate the two scoring functions in GOLD. For each of the 25 independent GA runs, a maximum number of 100000 GA functions were established on a set of five groups with a population size of 100 individuals. Mutation, migration and operator weights for crossover were set to 95, 10, and 95, respectively. Default cutoff values of 4.0 A for van der Waals distance and 2.5 A (dH-X) for hydrogen bonds were employed. When the top three solutions achieved RMSD values enough 1.5 A, GA docking was terminated. The RMSD values for the docking computations are based on the RMSD matrix of the ranked solutions and observed that the best ranked solutions were always among the first 50 GA runs, and further analyzing of the conformation of molecules performed on the best fitness score. The docking procedure was validated by redocking of tolrestat to the AKT1 crystal structure 3O96.
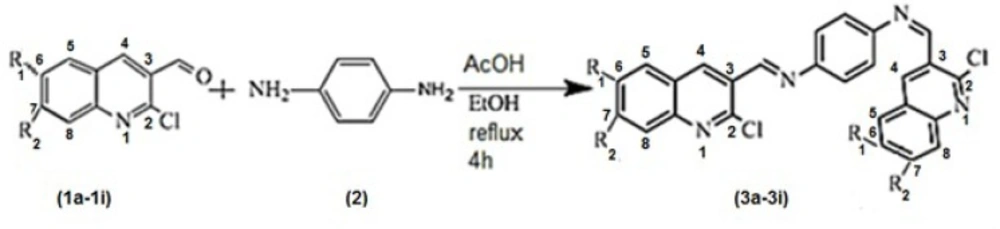
Figure 1
The best docked structure of 3b in the active site pocket of AKT1 (PDB entry: 3O96) in stick (left) and solvent surface (right) views.
Scheme1
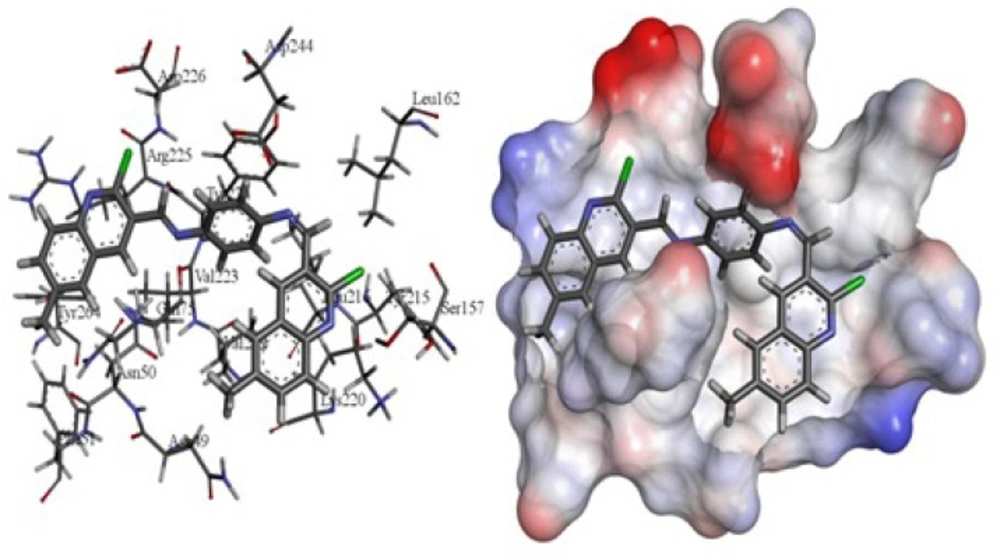
R1= H, Me, OMe, OEt, Cl, Et, Isopropyl R2= Me, Cl
Table1Synthesis of N1, N4-bis ((2-chloroquinolin-3-yl) methylene) benzene-1, 4-diamine derivatives
| mp °C | Yield (%)a | R | Product |
|---|---|---|---|
| 195 | 72 | H | 3a |
| 212-215 | 70 | 6-Me | 3b |
| 218-220 | 69 | 6-OMe | 3c |
| 196 | 68 | 6-OEt | 3d |
| 196-198 | 74 | 6-Cl | 3e |
| 198 | 70 | 7-Me | 3f |
| 190 | 69 | 6-Et | 3g |
| 200 | 68 | 6-Isopropyl | 3h |
| 195 | 75 | 7-Cl | 3i |
Table 2gold score, free energy of binding, Estimated inhibitory constant (Ki) and amino acids involved in hydrogen binding with synthetic compounds
| Residues involved in hydrogen binding | Ki | ∆G (kJ/mol) | Gold Score | compound |
|---|---|---|---|---|
| Lys 220, Trp 76 | 4.16817E-05 | -25 | 96.57 | 3a |
| Trp 76, Asn 49 | 1.65321E-06 | -33 | 113.76 | 3b |
| Tyr 224, Arg 225, Trp 76 | 1.51422E-05 | -27.51 | 98.19 | 3c |
| Tyr 224, Trp 76, Ile 80, Arg 225 | 1.15094E-05 | -28.19 | 107.58 | 3d |
| Trp 76, Ile 80, Arg 225 | 9.53038E-05 | -22.95 | 95.81 | 3e |
| Trp 76, Asn 49 | 2.86156E-06 | -31.64 | 100.98 | 3f |
| Lys 220, Asn 49, Trp 76 | 0.000113815 | -22.51 | 105.16 | 3g |
| Lys 220, Trp 76, Asn 49 | 3.56247E-09 | -48.22 | 104.44 | 3h |
| Trp 76, Tyr 224 | 3.39728E-08 | -42.63 | 102.05 | 3i |
| Tyr 76, Tyr 264, Arg 225, Lys 220 | 1.64349E-8 | -44.43 | 116.02 | Co crystall |
Results
Chemistry
To formation the products (3a-3i), the reaction took place between 2-chloroquinoline-3- carbaldehydes (1a-1i) and benzene-1, 4-diamine (2) as the starting materials in ethanol under reflux condition, Scheme1. The reaction was catalysed with acetic acid.
The completion of the reaction was monitored by TLC, and the disappearance of the starting material was observed within 4 h.
In order to study the generality of this method, we extended our studies to synthesis of some N1, N4-bis ((2-chloroquinolin-3-yl) methylene) benzene-1, 4-diamine derivatives (3a-3i). The reactions proceeded very efficiently in relatively high yields as showed in Table 1. All the products were characterized and confirmed by their spectroscopic and elemental analysis data.
a) Isolated yield
Docking
The receptor 3O96 is a complex structure of enzyme. The possible active site was identified using Accelrys DS Visualizer. Eight active site residues as Trp 80, Ile 84, Cer 205, Lys 268, Tyr 272, Ile 290, Asp 292 andCys 296 were found. Therefore it is chosen as a most biologically favorable site for docking. Some of the best models formed strong hydrogen bonds with Asn 49, Lys 220, Ser 157, Arg 225 and Trp 76 via quinoline moiety and nitrogen of quinolone ring (Figure 1.). pi - pi interaction between Lys 220, Trp 76, Tyr 224, Arg 225, Ile 80, and Asn 49 quinoline moiety was one of the common factor in enzyme-inhibitor junction. Amongst the synthetic compounds, 3e showed the lowest score while 3b possessed the highest score. The estimated inhibitory constant of the docked compounds are outlined in table.
Discussion
The complete spectral and elemental analytical data of the products confirmed the formation of new N1, N4-bis ((2-chloroquinolin-3-yl) methylene) benzene-1, 4-diamine derivatives (3a-3i). The 1H NMR spectrum of 3a consist of three singlet signals at δ 7.46, 9.09, 9.10 due to one aromatic and one imine hydrogen and one hydrogen of quinolone ring that posse nitrogen respectively. The doublet signal at δ 8.09 due to the hydrogen on the aromatic ring of quinolines and three multiple for the three hydrogens (on the aromatic ring of quinolines at δ 7.65, 7.84 and 8.10 ppm) were confirmed to the structure. The mass spectrum shows the molecular ion at 454 m/z. Also the absence of the stretching vibration band at 1689 cm-1 due to the carbonyl group (C=O) in IR spectrum confirmed the structure of 3a. The final proof for this structure received by the results of elemental analysis that was in good agreement calculated data.
Docking of N1, N4-bis ((2-chloroquinolin-3-yl) methylene) benzene-1, 4-diamine with AKT1 was performed by using of GoldScore fitness function. The algorithm exhaustively searches the entire rotational and translational space of the ligand with respect to the receptors. The various solutions valuated by a score, which is equivalent to the absolute value of the total energy of the ligand in the protein environment. For each compound the best docking solutions of GOLD score was considered. GoldScore carry out a force field based scoring function and is made up of four components: 1) Ligand internal van der Waals energy (internal vdw); 2) Ligand intermolecular hydrogen bond energy (internal-H-bond); 3) Protein- ligand hydrogen bond energy (external H-bond); 4) Protein-ligand van der Waals energy (external vdw). When the total fitness score is computed the external vdw score is multiplied by a factor of 1.375. This is an empirical correction to persuade protein-ligand hydrophobic interaction. The fitness function has been optimized for the divination of ligand binding positions. GoldScore = S (hb_ext) + S (vdw_ext) + S (hb_int) + S (vdw_int), where S (hb_ext) is the protein- ligand hydrogen bond score, S (vdw_ext) is the protein-ligand Van der Waals score, S (hb_int) is the score from intermolecular hydrogen bond in the ligand and S (vdw_int) is the score from intermolecular strain in the ligand. Redocking of 1-(1-(4-(7-phenyl-1H-imidazo (4, 5-g) quinoxalin- 6-yl) benzyl) piperidin-4-yl)-1H-benzo[d]imidazol- 2(3H)-one (co crystal) ∆G = -44.43 and Ki = 1.64E-8, bonding model of the mentioned molecule was similar to which was reported in the crystal structure of 3O96. It was noted that GOLD scores of 3b and 3d are 113.76 and 107.58, respectively, which are greater than the other scores as shown in Table 2.
Conclusion
In summary we have described an efficient and convenient synthesis of N1, N4-bis ((2-chloroquinolin-3-yl) methylene) benzene-1, 4-diamine involving condensation reaction of 2- chloroquinoline-3-carbaldehydes and p-phenylenediamine in acidic condition. Docking was carried out using GOLD 5.2. It was found that both hydrogen bonding and hydrophobic interaction play important roles in the structure and function of biological molecules, peculiarly for inhibition in a complex. It was noted that GOLD scores of 3b and 3d are greater than the other scores.
