Hyperglycemia appears to be an important factor in the development of diabetic neuropathy (
2). It induces neuronal damage and dysfunction probably via the generation of reactive oxygen species (
2,
39). As such, many studies have shown that HG does indeed cause neuronal cell death (-). However, there is conflicting evidence regarding the role of extracellular glucose during brain ischemic insults (
9). It is generally held that glucose supplies the essential energy needs of the brain and alteration of glucose metabolism may result in dysfunction of the CNS (
8,
14). A number of studies have obtained evidence for the beneficial effects of enhanced glucose entry into neurons during ischemia (
14,
40,
41). These works suggest a protective role for glucose against excitotoxicity and hypoxic ischemia. In the current study, we aimed to show the effects of HG on intracellular Ca
2+ levels, NO production, and general cell viability in the absence and presence of noscapine which we had previously shown to have neuroprotective effects of its own (
42).
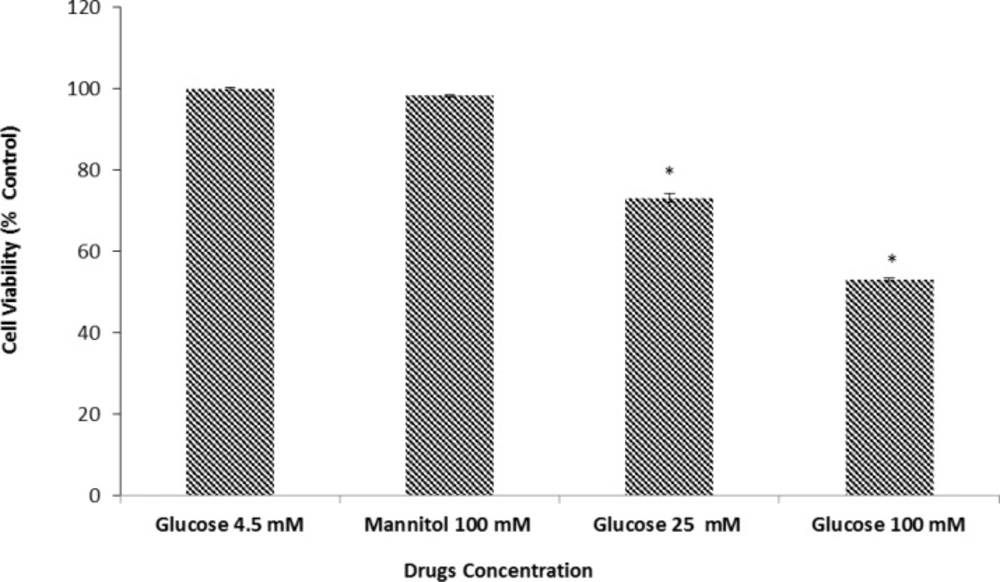
We examined the effect of D-glucose (25 or 100 mM) on neuronal cell death. Cortical cultures were exposed, for 24 h. to 4.5 mM (as normal glucose control), 25 and 100 mM glucose (as high glucose). Our results showed that 25 and 100 mM D-glucose could significantly reduce cell viability in a concentration dependent fashion as compared with control. In fact, neuronal cell viability treated with 100 mM glucose was markedly reduced as compared to 25 mM D-glucose. This was consistent with previous studies demonstrating that high concentrations of glucose induced neuronal injury in an
in-vitro model (
2,
4,
43). For example, Koshimura K, Tanaka J, Murakami Y and Kato Y () showed that HG increased NO production and induced cell death in differentiated PC12 cells. Also, Vincent AM, McLean LL, Backus C and Feldman EL () demonstrated that in neurons, HG caused oxidative stress, induced mitochondrial dysfunction and programmed cell death. Thus, the data obtained by us and other workers, confirmed exposure to HG for 24 h. or more, to be detrimental to neuronal cells.
However, there is data to suggest a neuroprotective role for short period exposure to HG after ischemia (
40). Here the mechanism of protection is not completely delineated. Toping up of intracellular glucose stores has been proposed to be the reason for this effect (
9,
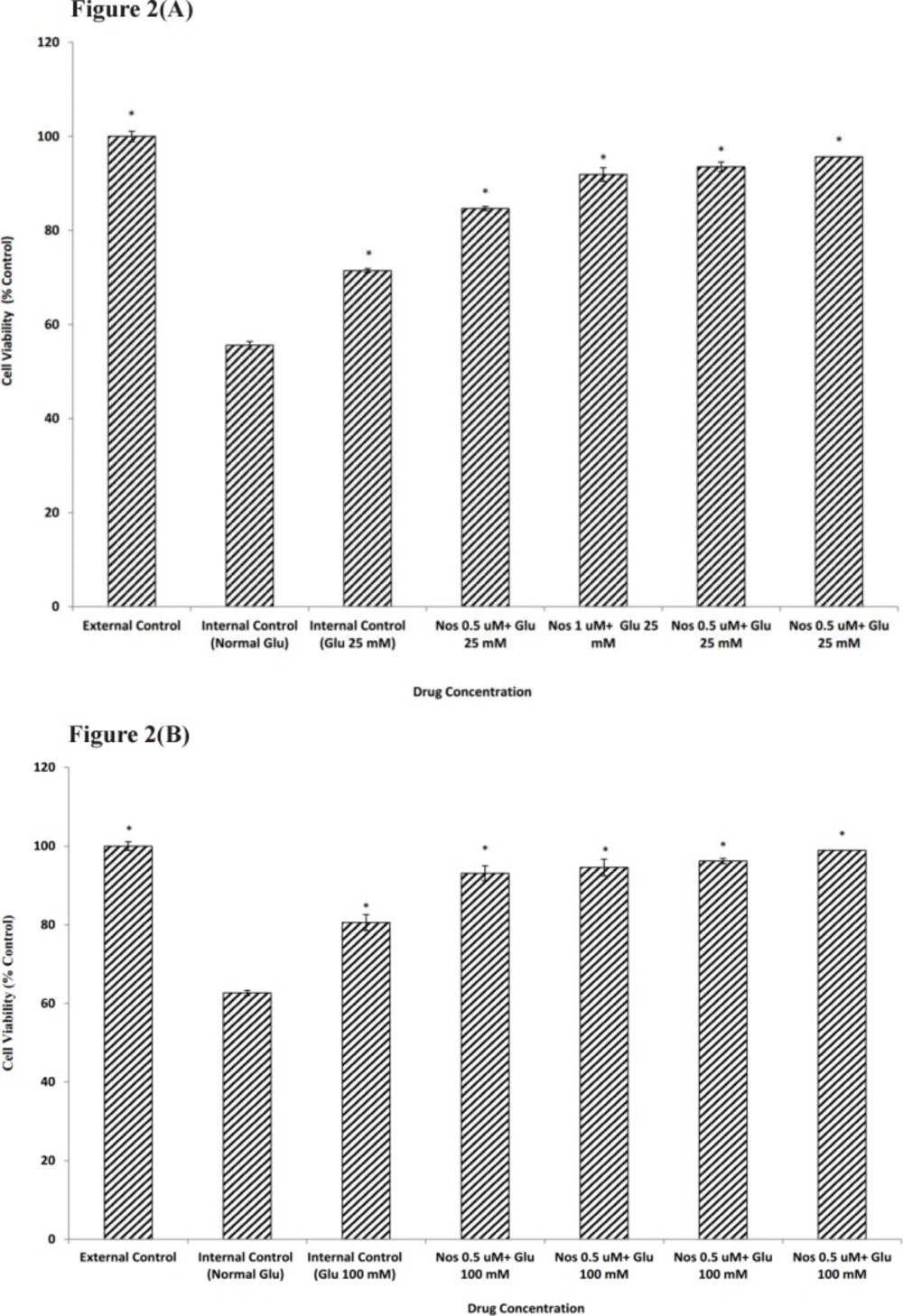
44). Thus, to determine whether a short exposure to HG could protect cortical neuronal cells from OGD insult, cells were pre-incubated for 24 h. in RPMI-1640 containing 25 or 100 mM glucose prior to 30 min OGD. Exposure to HG reduced cell death due to OGD. This effect was more visible for 100 mM D-glucose than that of 25 mM D-glucose.
Other workers have shown HG to be protective against the effects of excitotoxins (
8,
9,
41). Seo SY, Kim EY, Kim H and Gwag BJ () showed that although pretreatment with 25 mM glucose did not induce great protection against NMDA evoked neuronal death, 100 mM glucose pretreatment enhanced cell survival after treatment with NMDA. It has been demonstrated that OGD induces cell death by enhancing the release of extracellular glutamate which in turn increases activation of NMDA receptors leading to increased intracellular Ca
2+ levels and augmented NO production (-). Thus the data obtained by us is in part agreement with the work of many other researchers (
8,
9,
14,
40,
41).
All this suggests an important role for intracellular Ca2+ and NO in both excitotoxicity and OGD induced cell death.
In a previous study carried out in this lab, effects of noscapine on OGD induced death in primary cortical neurons had been studied (
42). Noscapine appeared to impart protection against ischemia by attenuating OGD induced NO production. In this study we investigated the possibility that noscapine could potentiate the protective effects of HG from OGD in primary cortical neurons. When neuronal cells pre-treated with noscapine (0.5-2 µM) subsequent the addition of HG ( 25 or 100 mM) and then subjected to 30 min OGD, the cell viability increased as compared with the cell pre-treated with high-glucose alone. In order to find out the neuroprotective effect of noscapine in HG condition, we set out to measure the change in Ca
2+ and NO after OGD in cells pretreated with HG.
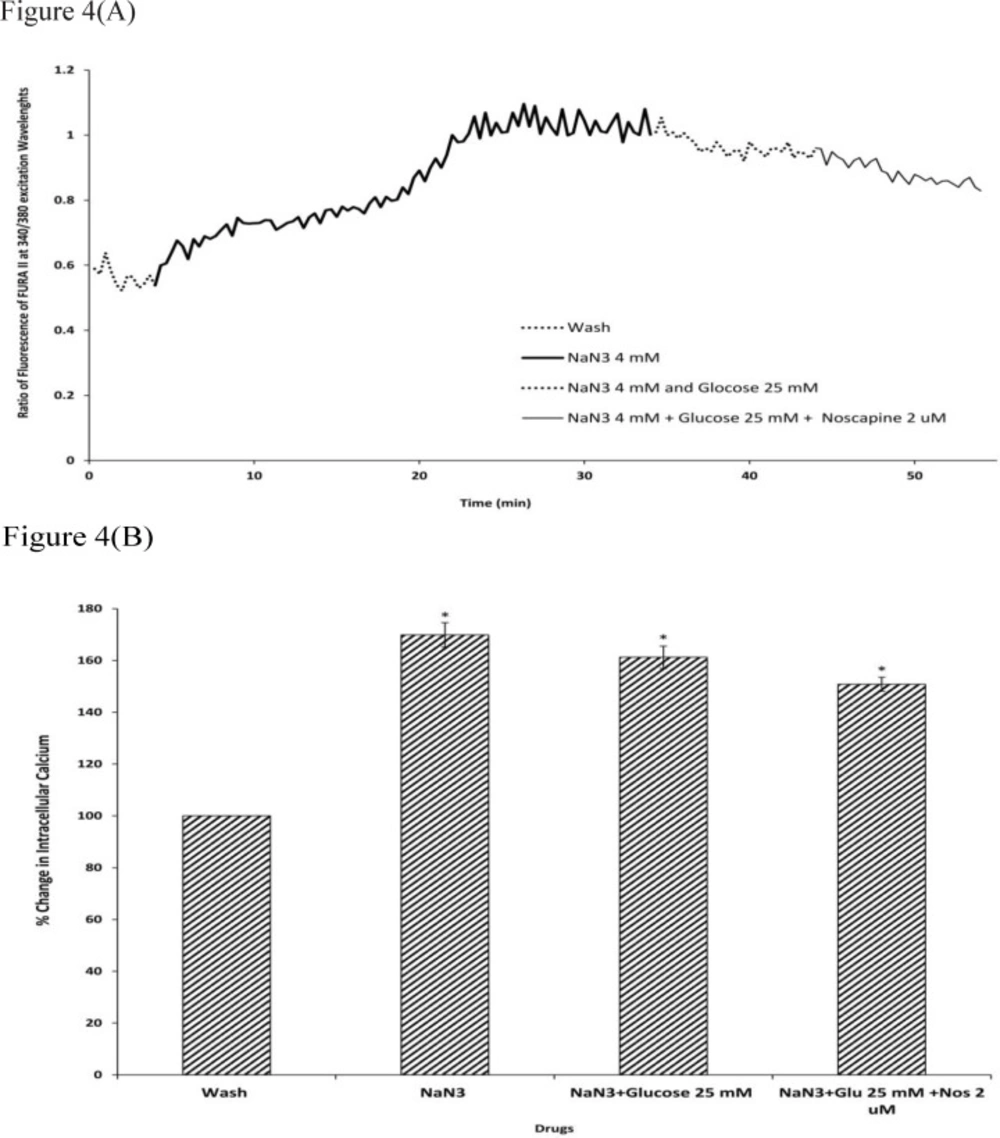
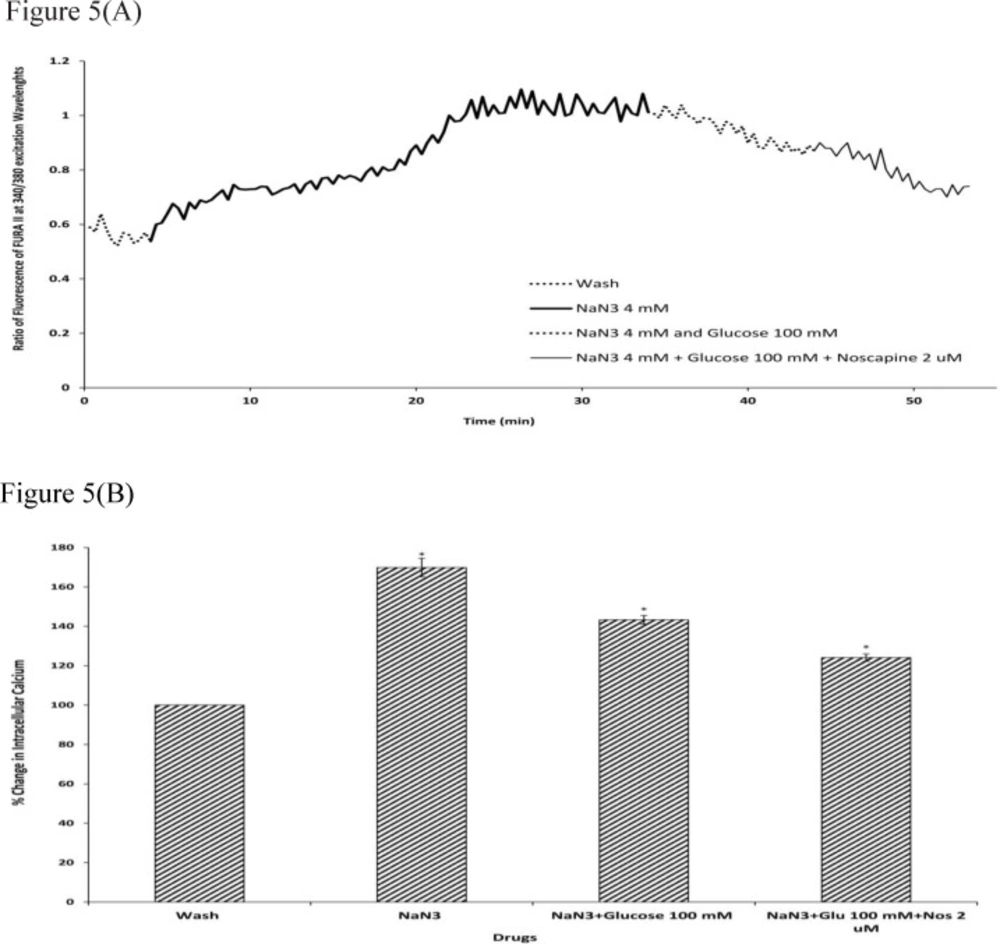
Sodium azide/glucose deprivation (chemical OGD) model was used to create
in-vitro ischemic conditions. This model has been shown to elicit electrophysiological and neurochemical changes that are similar to the OGD model of ischemia. While this model having the advantage of producing neurochemical changes more rapidly and reproducibly (
49,
50), thus facilitating the recording of changes in (Ca
2+)i (
51). Our results showed that during chemical OGD the (Ca
2+)i was significantly increased compared to control condition. This is consistent with previous studies indicating that NaN
3 could increase neuronal (Ca
2+)i, as a consequence of energy depletion (
52).
We next examined the (Ca2+)i level in neuronal cells exposed to 25 or 100 mM D-glucose after 30 min of chemical OGD treatment. The results suggested that exposure to 25 or 100 mM D-glucose was able to reverse the effects of chemical OGD on intracellular Ca2+ levels in cultured cortical neurons. Also, 100 mM glucose was more effective in decreasing (Ca2+)i levels than 25 mM glucose. Chan J and Greenberg DA () showed that in PC12 cells, increasing glucose concentration in the culture medium by a factor of 4, attenuated ischemic injury by reducing the dihydropyridine-sensitive and depolarization-induced increase in Ca2+. These results suggested that HG might decrease neuronal cell death by reducing (Ca2+)i levels in neuronal cells.
Finally, our findings demonstrated that cells exposed to D-glucose together with 2 µM noscapine following chemical OGD had significantly lower (Ca2+)i compared to D-glucose treatment alone. This data suggests that noscapine reinforced the protective effect of HG via reduction in intracellular Ca2+ levels. Also, noscapine potentiated the effects of 100 mM glucose more than it decreased the effects of 25 mM glucose. However, because 100 mM D-glucose could considerably reduce cell death during ischemic insult, the neuroprotective effect of 2 µM noscapine in presence of 100 mM D-glucose also could better act on neuronal cells to reduce (Ca2+)i following chemical OGD.
Many studies have been shown that NO is involved in both physiological and pathological events, and is believed to play different roles in both lethal and sub-lethal events (
47,
54). Several studies have found evidence indicating an involvement of NO synthase (NOS) in glutamate-induced excitotoxicity and there were some corroborating data that NO production increases during ischemia (
35,
45,
47, -). For example, it has been shown that during OGD, calcium enters the neurons through NMDA receptor and/or L-type voltage-gated calcium channel activation and activates NOS which leads to NO production (
45). Zhang M, Ning G-M, Hong D-H, Yang Y, Kutor J and Zheng X-X () had shown that Ca
2+-free medium markedly inhibited the OGD-induced increase in NO, suggesting that the inhibition of Ca
2+ might cause the decrease of NO synthesis.
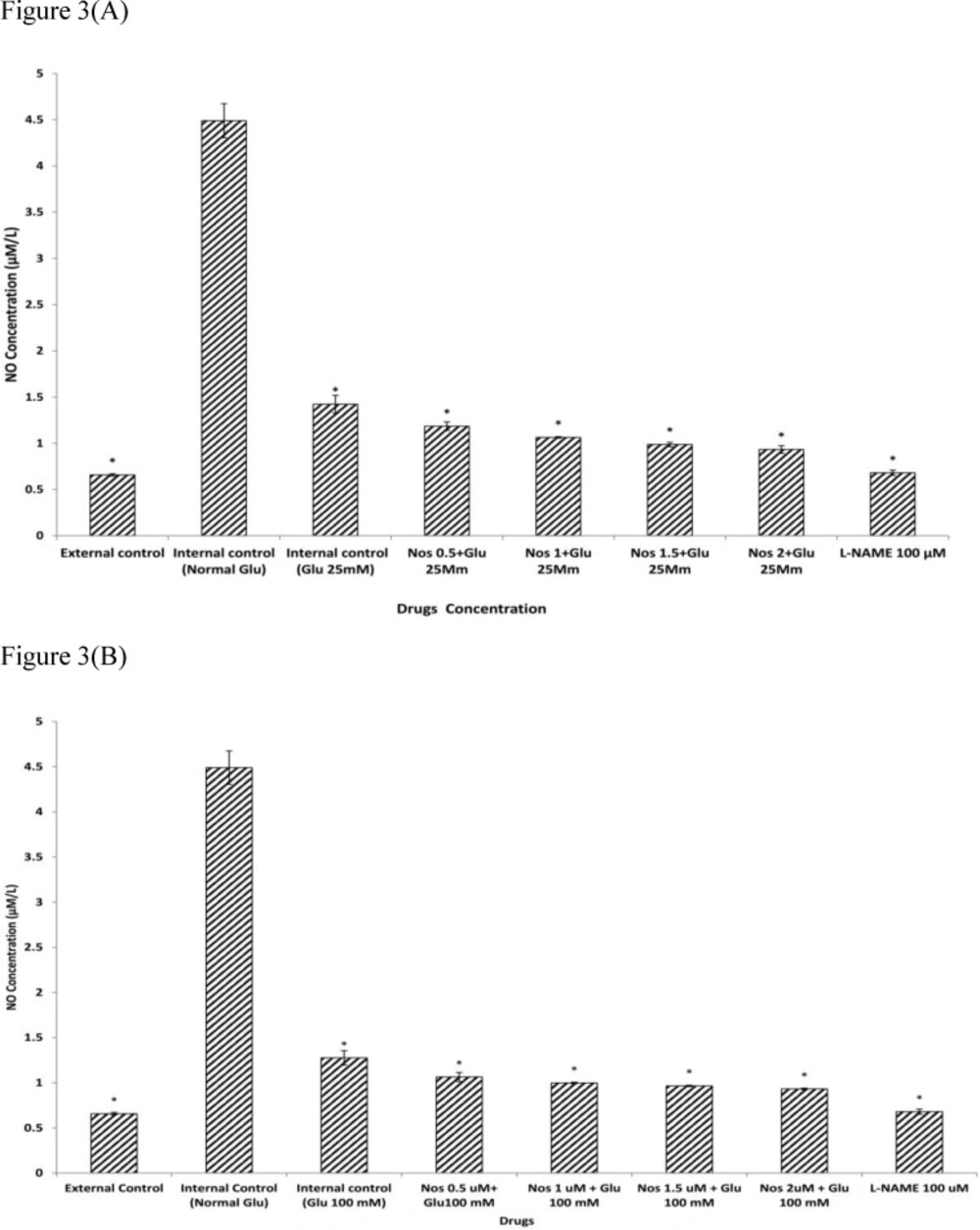
Our results confirmed that in neuronal cells exposed to 30 min OGD, NO production was significantly increased, supporting a role for NO in the cascade of events leading to OGD-induced cell death (
45). However, the exposure of neuronal cells to 25 or 100 mM D-glucose could decrease the NO production during 30 min OGD. After that neuronal cells were pre-treated with noscapine (0.5-2 µM) in the presence of the 25 or 100 mM D-glucose and then subjected to 30 min OGD. The results indicated that the NO production was significantly reduced as compared with 25 or 100 mM D-glucose alone. This suggested that noscapine also increased neuroprotective effect of HG by reducing NO production. In addition, when the cultures were pre-treated with the NOS inhibitor, L-NAME, NO production was significantly decreased and returned to the level of the external control. Suggesting that NOS inhibitors could markedly decrease the NO production after NMDA receptor activation, and decreased excitotoxic injury in cultured neurons (
45,
58,
59)
In conclusion, noscapine potentiated the neuroprotective effect of HG (25 and 100 mM) treatment prior to OGD. The enhanced cell viability after treatment with both noscapine and HG was mirrored by a dampening of OGD induced intracellular Ca2+ and NO rise. These observations support the hypothesis that calcium homeostasis and NO production have crucial roles in excitotoxicity process.