Materials
Telmisartan (TEL) was obtained from Eurapharm, Inc. (Suwon, South Korea). Polyvinylpyrrolidone K30 (PVP K30, Povidone® K30) was supplied by BASF (Shanghai, China) and the polyethylene glycol 6000 (PEG 6000) was purchased from Shinyo (Osaka, Japan). Sodium hydroxide (NaOH) was purchased from Sinopharm Chemical Reagent Co. (Shanghai, China). Poloxamer 188 (Lutrol® F68) was kindly provided by Shanghai Chineway Pharm. Tech. Co. (Shanghai, China). All other reagents and chemicals were of analytical grade and used without further purification.
Preparation of organic solvent-free solid dispersions (OSF- SDs)
The OSF-SDs containing TEL were prepared by a lyophilization method as reported in the literature (
15). The detailed formulation compositions are shown in
Table 1. Briefly, NaOH was dissolved in distilled water and formed an alkaline solution. TEL and PVP K30 (or PEG 6000) were accurately weighed and dissolved in the alkaline solution with a continuous stirring for the preparation of OSF-SD1 and OSF-SD3. On the other hand, TEL, PVP K30 (or PEG 6000) and poloxamer 188 were also dissolved in the upper alkaline solution for the preparation of OSF-SD2 and OSF-SD4. The resulting solutions were then frozen at -70 °C and lyophilized using a lyophilizer (Christ Alpha 1-4, Germany). After the lyophilization, the solid masses were grinded and sieved to obtain a particle size fraction of 125–500µm.
| Formulation code | TEL(g) | PVP K30 (g) | PEG 6000 (g) | Poloxamer188 (g) | NaOH(g) | Distilled water(mL) |
|---|
| OSF-SD1 | 0.8 | 2.4 | - | - | 0.08 | 4 |
| OSF-SD2 | 0.8 | 2.4 | - | 0.16 | 0.08 | 4 |
| OSF-SD3 | 0.8 | - | 2.4 | - | 0.08 | 4 |
| OSF-SD4 | 0.8 | - | 2.4 | 0.16 | 0.08 | 4 |
Analysis of drug content
The OSF-SDs equivalent to 80 mg of TEL loading were accurately weighted and dissolved in 100 mL of methanol. One milliliter of above solution was diluted into 10 mL, and then filtered through 0.45 µm Millipore filters. The TEL in filtrate was analyzed by a HPLC method described below.
Scanning electron microscopy (SEM)
The OSF-SDs were detected by S-4700 (Hitachi, Tokyo, Japan) scanning electron microscope in high vacuum mode. Samples were coated with gold for 180 s using a JEOL JFC-1100 sputter coater (Jeol, Tokyo, Japan) under argon atmosphere and then imaged at ambient temperature at 15 kV and observed at magnifications of 100×.
Differential scanning calorimetry (DSC)
DSC measurements were performed using a differential scanning calorimeter (Model 2010, TA Instruments, USA). Accurately weighed samples (20-50 mg) including raw material, excipients, OSF-SDs and their physical mixtures (PMs) were sealed in aluminum pans. An empty aluminum pan was used as reference. The samples were heated at a scanning rate of 10 °C/min from 50 to 300 °C under a dry nitrogen gas purge.
X-ray powder diffractometry (XRD)
The XRD patterns of the raw material, excipients, OSF-SDs and their PMs were recorded by using an X-ray diffractometer (MERCURY CCD, Japan) with tube anode Cu over the interval 5–60°/2θ. The scanning rate was adjusted to 2°/min.
Fourier transform-infrared (FT-IR) spectroscopy
The spectra of the samples including the raw material, excipients, OSF-SDs and their PMs were characterized using a FTIR spectrophotometer (Model Excaliber Series UMA-500, Bio-Rad, USA). KBr pellets were prepared by gently mixing 1 mg of the sample with 200 mg KBr. The wavelength ranged from 400 to 4,000 cm-1 with a resolution of 2 cm-1.
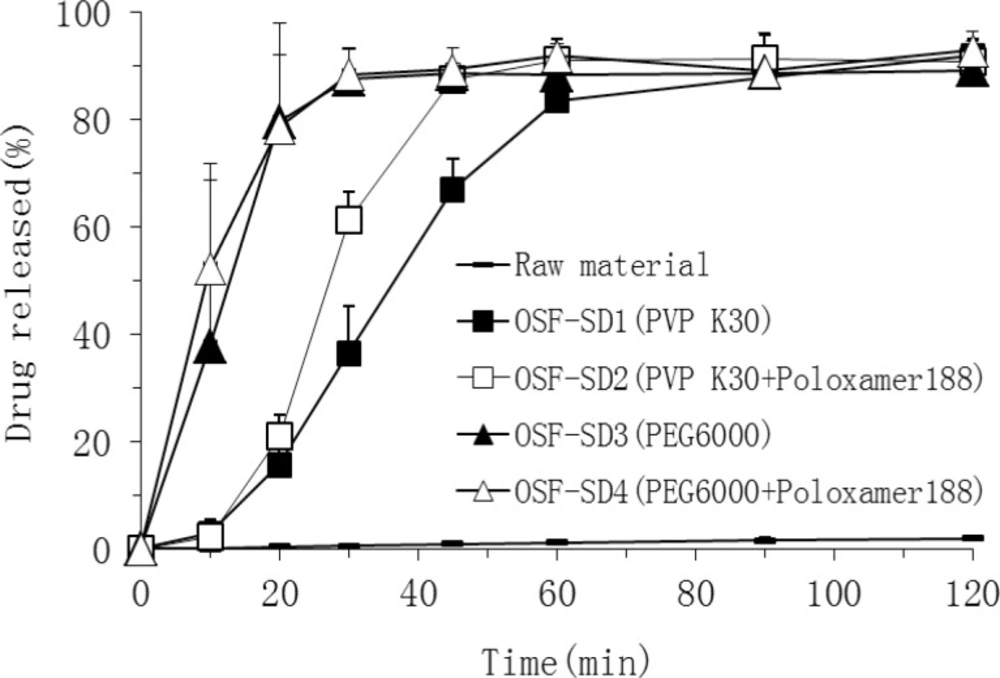
Dissolution studies
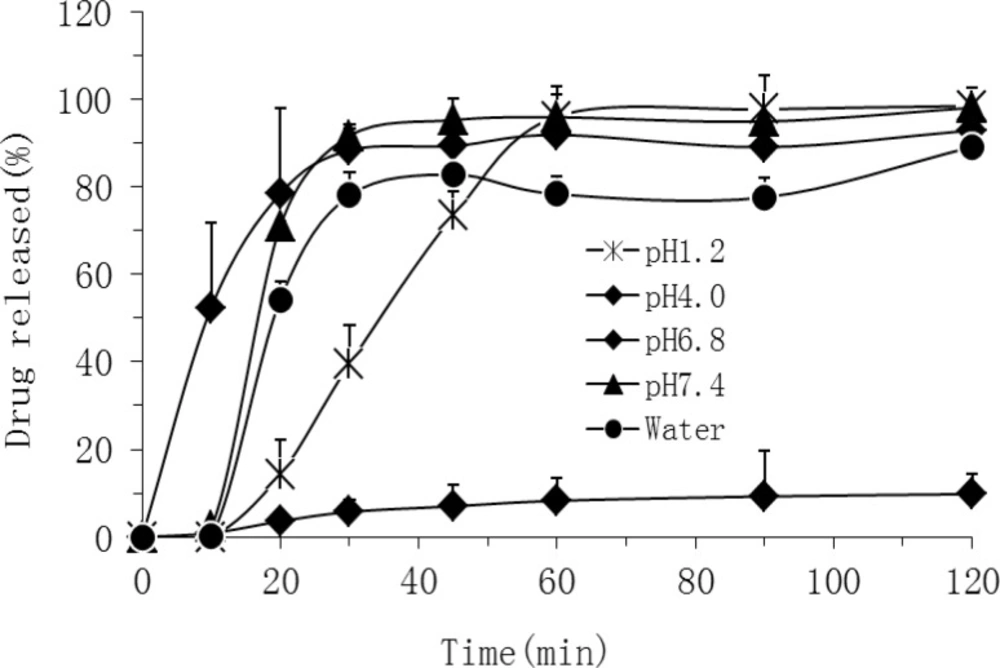
Dissolution studies were performed for both TEL bulk powder and OSF-SDs equivalent to 80 mg of TEL loading according to the USP dissolution I basket method at a rotation speed of 75 rpm in the 900 mL of pH 6.8 phosphate buffer solution at 37±0.5 °C (
16). At predetermined intervals, 5 mL of the samples was withdrawn and filtered through a membrane filter (0.45μm). The concentration of TEL was assayed by a HPLC method described below. All experiments were performed in triplicate. In addition, dissolution studies were also carried out in the media with different pH values (pH1.2, pH4.5, pH7.4) and water.
High performance liquid chromatography (HPLC) analysis
TEL was analyzed using a HPLC system (Shimadzu, LC-20A, Japan) consisting of a LC-20AD pump and a SPD-M20A detector. Analysis was carried out on a Phenomenex C
18 column (250 × 4.6 mm, 5 μm). The mobile phase consisted of a 60 : 40 (%v/v, pH3.7) mixture of acetonitrile and 10 mM potassium dihydrogen phosphate. The flow rate was 1.0 mL/min and the column temperature was maintained at 35 °C. The injection volume was 20 μL and the detection wavelength was 296 nm (
4).
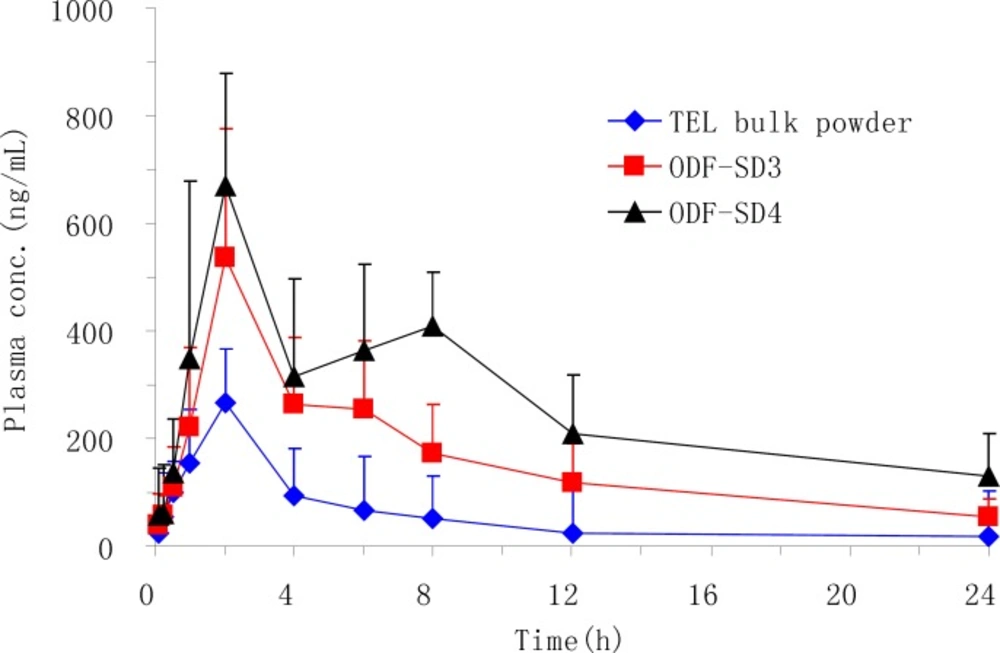
Bioavailability study
Animal study
Before beginning any experiment, we obtained approval for this study from the Institutional Review Board on Animal Research and Ethics Committees of Soochow University. Eighteen male SD rats weighing 260–320 g were randomly divided into three treatment groups, each consists of six rats. The rats were fasted over 12 h prior to the experiments. A polyethylene cannula (inner diameter, 0.58 mm; outer diameter 0.96 mm) was surgically introduced into the left femoral artery of rat under ether anesthesia to obtain blood samples at the various sampling times. The mini capsules containing TEL bulk powder, OSF-SD3 and OSF-SD4 were administered orally at a dose equivalent to 3.6mg (TEL)/kg body weight using an oral sonde to three groups respectively. Approximately 0.5 mL of blood sample was collected in a heparinized tube using an indwelling cannula at 0.15, 0.5, 1, 2, 4, 6, 8, 12 and 24 h, and then centrifuged at 1500 × g for 10 min to collect the plasma samples. The plasma samples were stored in a freezer at −40 °C until being analyzed by HPLC (17).
Preparation of plasma sample
Twenty microliters of itraconazole (4 μg/mL) as an internal standard were added to 200 μL of each plasma sample and voltexed for 5 s. Fifty microliters of 10 % phosphoric acid and 1 mL of diethyl ether were added to the mixture and then agitated for 5 min using a vortex mixer. Thereafter, the mixture was centrifuged at 1500 × g for 10 min. The organic phase was transferred into a tube and evaporated under a gentle stream of nitrogen. The residue was then reconstituted with 50 μL of methanol and 20 μL of the resulting solution was injected into the HPLC system.
HPLC analysis for plasma samples
Chromatographic separation was performed at a flow rate of 1.0 mL/min, at a wavelength of 254 nm, using a Phenomenex C18 (4.6 × 250 mm, 5 μm) column. The column temperature was maintained at 35 °C. The mobile phase was acetonitrile: 0.01mol/L phosphate buffer (60:40, v/v, pH 3.7) (
4).
Data analysis
Pharmacokinetic parameters of TEL were calculated using non-compartmental methods. The maximum plasma concentration (Cmax) and the time to reach the Cmax (Tmax) were read directly from the plasma concentration–time profiles of TEL. The areas under the plasma concentration–time curve from zero to 24 h (AUC0–24h) were calculated using the classical trapezoidal method. All data are presented as mean ± standard deviation. The statistical significance of the differences was performed using an analysis of variance (ANOVA) test and a p value < 0.05 or 0.01 was considered significant.


![DSC thermograms of TEL (A), PVP K30 (B), PEG 6000 (C), NaOH (D), poloxamer 188 (E), OSF-SDs [OSF-SD1 (F), OSF-SD2 (G), OSF-SD3 (H), OSF-SD4 (I)] and their physical mixtures [PM1 (J), PM2 (K), PM3 (L), PM4 (M)].](https://brieflands.com/journals/ijpr/articles/125187/figures/ijpr-15-385-g002-preview.webp)
![XRD patterns of TEL (A), PVP K30 (B), PEG 6000 (C), NaOH (D), poloxamer 188 (E), OSF-SDs [OSF-SD1 (F), OSF-SD2 (G), OSF-SD3 (H), OSF-SD4 (I)] and their physical mixtures [PM1 (J), PM2 (K), PM3 (L), PM4 (M)].](https://brieflands.com/journals/ijpr/articles/125187/figures/ijpr-15-385-g003-preview.webp)
![FT-IR spectra of TEL (A), PVP K30 (B), PEG 6000 (C), NaOH (D), poloxamer 188 (E) and OSF-SDs [OSF-SD1 (F), OSF-SD2 (G), OSF-SD3 (H) and OSF-SD4 (I)].](https://brieflands.com/journals/ijpr/articles/125187/figures/ijpr-15-385-g004-preview.webp)