
Introduction
Monoamine oxidases are widely distributed enzymes that contain a flavin adenine dinucleotide (FAD) covalently bounded to a cysteine residue (1). Many living organisms possess MAOs and in mammals two isoforms are present, MAO-A and MAO-B, located in the outer membrane of the mitochondria. These two isoforms are involved in the oxidative deamination of exogenous and endogenous amines, including neurotransmitters, thus modulating their concentrations in the brain and peripheral tissues. Physiologically, MAOs oxidize biogenic neurotransmitters such as dopamine, norepinephrine, 5-hydroxytryptamine (5-HT, serotonin) and β-phenethylamine, dietary, and xenobiotic amines such as tyramine and benzylamine (2, 3). MAO-A inhibitors are frequently used as antidepressants and anti-anxiety agents while MAO-B inhibitors, alone or combined with L-Dopa, are relevant tools in the therapy of Alzheimer’s and Parkinson’s diseases (4). Development of MAO inhibitors is important not only from the standpoint of symptomatic treatment, but also with regard to the neuroprotective effects (5).
The rhizomes of Rheum palmatum, named “Jun Za” in traditional Chinese-Tibetan Medicine, have been used as medicament of astriction, choleplania and pyrexia in Tibet, China for thousands of years (6). In the searching for naturally occurring MAO inhibitors from plant, we found that the extract of Rheum palmatum showed potential MAO inhibition with IC50 of 10.49 μg/mL (7). Further bioactivity-guided fractionation resulted in the isolation of seven stilbenes and one catechin. Herein the report, isolation, structure elucidation and MAO inhibitory property of these compounds is demonstrated.
Material and Methods
General experimental procedures
NMR spectra were recorded with a Varian Mercury-400BB NMR spectrometer. Semi preparative HPLC system consisted of a Jasco PU-2086 Plus, UV -2075 Plus detector and YMC-Pack ODS-A column (5μM, φ 250 × 10 mm, YMC Co. Ltd.). Silica gel 200-300 mesh for column chromatography and silica GF254 for TLC were supplied by the Qingdao Marine Chemical Inc., China. MCI-GEL CHP20 (75-150μM) were from Mitsubishi Chemical Holdings Corp. Macroporous resin (HPD-100) were purchased from Cangzhou Baoen Chemical Inc., China. Polyamide 100-200 mesh for column chromatography was purchased from Taizhou Luqiao Sijia Biochemical Plastic Factory, China.
Plant material
Rhizomes of Rheum palmatum were purchased from Bayi Herb Market in Xining, China, which were identified by Associate Professor, Lin Yang who majored in plant classification, School of Life Science and Engineering, Lanzhou University of Technology, Lanzhou, China. A voucher specimen (No. 2011071719) is deposited at the School of Life Science and Engineering, Lanzhou University of Technology.
Extraction and isolation
Dried rhizomes of Rheum palmatum (2.4 Kg) were powdered and refluxed with EtOH (15 L×2) for 2 h. The EtOH extract was evaporated in vacuo, yielding a extractum. To remove alkaline constituent, the extractum was suspended with distilled water (2 L) and adjusted pH to 10-11, then filtered. The filtrate was adjusted pH to 2-3, and extracted with the EtOAc 3 times. The EtOAc extract liquor was evaporated in vacuo, yielding a extractum (80 g). The extractum was suffered on pre-fractionation using macroporous resin (HPD-100, Cangzhou Bon Absorber Technology Co. Ltd., China) (5 Kg) with a step gradient of EtOH-H2O solvent system (0:100, 30:70, 50:50, 70:30, 100:0, v/v) as an eluent to yield 6 fractions (A1~A6). A3 (16 g) was chromatographed on polyamide (100-200 mesh, 300 g) using a step gradient of MeOH-H2O solvent system (0:100, 30:70, 50:50, 70:30, 100:0, v/v) as an eluent to yield 6 fractions (A3-1~A3-6). A3-5 (2 g) was re-chromatographed on MCI gel (200 g) with a stepwise gradient of MeOH-H2O solvent system (0:100, 30:70, 50:50, 70:30, 100:0, v/v) as an eluent to yield 6 fractions (A3-5-1~A3-5-6). A3-5-4 (350 mg) was separated successively on silica gel (200-400 mesh, 350 g) with a stepwise gradient of CHCl3 and MeOH (20:1, 15:1, 10:1, 5:1, 1:1, v/v) to produce compounds 4 (130 mg) and 2 (10 mg). A3-5-3 (50 mg) was purified by polyamide (100-200 mesh, 50 g) using MeOH-H2O (50:50, v/v) as a solvent system to give 6 (15 mg). A3-5-6 (30 mg) was re-crystallized with MeOH to yield 1 (20 mg). A5 (15 g) was purified by silica gel (200-400 mesh,150 g) with a stepwise gradient of CHCl3 and MeOH (20:1, 15:1, 10:1, 5:1, 1:1, v/v) as an eluent to yield 5 fractions (A5-1~A5-5), A5-5 (1 g) was purified by silica gel(200-400 mesh, 150 g) with a stepwise gradient of petroleum ether-acetone(10:1-1:1, v/v) to produce 8 (30 mg). A1(2 g) was chromatographed on silica gel (200-400 mesh, 200 g) with a stepwise gradient of CHCl3 and MeOH (10:1, 8:1, 5:1, 1:1, v/v) to produce 7 (10 mg). A1-1 (40 mg) and A1-2 (100 mg) were separated successively by semi-preparative HPLC (YMC-Pack ODS-A, 250 × 10 mm, 5 μM, flow rate 2 mL/min) eluting with MeOH-H2O (40:60, v/v) to produce 5 (15 mg, tR 28 min) and 3 (10 mg, tR 43 min), respectively.
Piceatannol 1
Light yellow powder, C14H12O4. ESI-MS m/z:243[M-H]+. 1H-NMR (400 MHz, CD3COCD3): δ ppm 7.07 (1H, d, J = 2.0 Hz, H-2'), 6.95 (1H, d, J = 16.0 Hz, H-β), 6.90 (1H, dd, J = 8.0 , 1.2 Hz, H-6'), 6.80 (2H, m, H-α, 5'), 6.53(2H, s, H-2,6), 6.26 (1H, s, H-4). 13C-NMR (100 MHz, CD3COCD3): δ 159.8 (C-3, 5), 146.6 (C-3'), 141.4 (C-1), 131.2 (C-1'), 129.8 (C-β), 127.1 (C-α), 120.3 (C-6'), 120.3 (C-6'), 116.6 (C-5'), 114.0 (C-2'), 105.9 (C-2,6), 102.8 (C-4). (8)
Resveratrol 2
White powder, C14H12O3. 1H-NMR (400 MHz, CD3COCD3): δ ppm7.42 (2H, d, J = 8.4Hz, H-2', 6'), 7.02 (1H, d, J = 16.6 Hz, H-β), 6.88 (1H, d, J = 16.6 Hz, H-α), 6.84 (2H, d, J = 8.3 Hz, H-3', 5'), 6.54 (2H, d, J = 2.0 Hz, H-2, 6), 6.27 (1H, d, J = 2.0 Hz, H-4). 13C-NMR (100 MHz, CD3COCD3): δ ppm 159.6 (C-3, 5), 158.2 (C-4'), 141.0 (C-1), 130.0 (C-1'), 129.2 (C-β), 128.8 (C-2', 6'), 126.9 (C-α), 116.5 (C-3', 5'), 105.8 (C-2, 6), 102.7 (C-4) (9).
Piceid 3
Amorphous powder, C20H23O8. 1H-NMR (400 MHz, CD3OD): δ ppm 7.35 (2H, d, J = 8.0 Hz, H-2', 6'), 7.02 (1H, d, J = 16.0 Hz, H-β), 6.83 (1H, d, J = 16.0 Hz, H-α), 6.78 (3H, m, H-2, 3', 5'), 6.60 (1H, brs, H-6), 6.43 (1H, brs, H-4), 4.90 (1H, d, J = 7.2 Hz, H-1''), 3.30–3.47 (4H, m, H-2'', 3'', 4'' and 5''), 3.70 (1H, dd, J = 11.0, 5.4 Hz, H-6b''), 3.90 (1H, dd, J = 11.0, 1.5 Hz, H-6a'') (10).
Rhapontigenin 4
Yellow amorphous powder, C20H22O9.1H-NMR (400 MHz, CD3COCD3): δ ppm δ7.09 (1H, brs, H-2'), 6.86-6.99 (4H, m, H-5', H-6', H-β, H-α), 6.55 (2H, brs, H-2, 6), 6.27 (1H, brs, H-4), 3.84 (3H, s, 4'-OCH3). 13C-NMR (100 MHz, CD3COCD3): δ ppm 159.5 (C-3, 5), 148.3 (C-3'), 147.6 (C-4'), 140.7 (C-1), 131.7 (C-1'), 129.1 (C-β), 127.6 (C-α), 119.7 (C-2'), 113.2 (C-5'), 112.3 (C-6'), 105.7 (C-2, 6), 102.7 (C-4), 56.2 (4'-OCH3) (11).
Piceatannol-3'-O-β-D- glucopyranoside 5
Yellow amorphous powder, C20H22O9. 1H-NMR (400 MHz, CD3OD): δ ppm 7.46 (1H, d, brs, H-2'), 7.05 (1H, dd, J = 2.0, 8.6 Hz, H-6'), 6.92 (1H, d, J = 16.4 Hz, H-α), 6.81 (2H, m, H-β, 5'), 6.45 (2H, d, brs, H-2, 6), 6.16 (1H, brs, H-4), 4.80 (1H, d, J = 7.6 Hz, H-1''), 3.94 (1H, J = 11.0, 1.5 Hz, H-6a''), 3.72 (1H, dd, J = 11.0, 5.4 Hz, H-6b''), 3.30-3.52 (4H, m, H-2'' toH-5''). 13C-NMR (400 MHz, CD3OD) δ ppm: 159.6 (C-3, 5), 148.4 (C-3'), 147.1 (C-4'), 141.1 (C-1), 131.2 (C-1'), 129.2 (C-α),127.7 (C-β), 123.6 (C-6'), 117.2 (C-5'), 116.6 (C-2'), 105.8(C-2, 6), 104.5 (C-4), 102.7 (C-1''), 78.6 (C-5''), 77.8 (C-2''), 75.1 (C-3''), 71.6 (C-4''), 62.7 (C-6'') (8).
Rhaponticin 6
Light yellow amorphous powder, C15H14O4. 1H-NMR (400 MHz, CD3OD): δ ppm 7.00 (1H, d, brs, H-2'), 6.81-6.98 (5H, m, H-2, 5', 6', α, β), 6.61 (1H, brs, H-2), 6.45 (1H, brs, H-4), 3.82 (3H, s, 4'-OCH3), 4.88 (1H, d, J = 7.6 Hz, H-1''), 3.92(1H, J = 11.0, 2.0 Hz, Glc H-6a''), 3.70 (1H, dd, J = 11.0, 5.6 Hz, Glc H-6b''), 3.35-3.50 (4H, m, H-2'' toH-5''). 13C-NMR (100 MHz, CD3OD): δ ppm 160.4(C-5), 159.6 (C-3), 149.1 (C-4'), 147.9 (C-3'), 141.3 (C-1), 132.4 (C-1'),129.5 (C-α), 128.0 (C-β), 120.2 (C-6'), 113.8 (C-2'), 112.9(C-5'), 108.3 (C-6), 107.0 (C-2), 104.1 (C-4), 102.4 (C-1''), 78.2 (C-5''), 78.0 (C-3''), 74.9 (C-2''), 71.5 (C-4''), 62.6 (C-6''), 56.5 (4'-OCH3) (11).
Catechin 7
Yellow amorphous powder, C15H13O6. 1H-NMR (CD3OD, 400 MHz): δ ppm 6.83 (1H, d, J = 1.8 Hz, H-2'), 6.75 (1H, d, J = 8.2 Hz, H-5'), 6.71 (1H, dd, J = 8.1,1.8 Hz, H-60), 5.92 (1H, br s, H-8), 5.85 (1H, d, J = 2.2 Hz, H-6), 4.56 (1H, d, J = 7.4 Hz, H-2), 3.97 (1H, m, H-3), 2.84 (1H, dd, J = 16.0, 5.2 Hz, H-4a), 2.50 (1H,dd, J = 16.1, 8.1 Hz, H-4b) (12).
Desoxyrhapontigenin 8
White powder, C15H14O3. 1H-NMR (CD3OD,400 MHz): δ ppm 7.41 (2H, d, J = 8.8 Hz, H-2', 6'), 6.97 (1H, d, J = 16.0 Hz, H-α), 6.81-6.88 (3H, m, H-3', 5', β), 6.46 (2H, brs, H-2, 6), 6.16 (1H, brs, H-4), 3.77 (3H, s, 4'-OCH3). 13C-NMR (100 MHz, CD3OD): δ ppm: 160.9 (C-4'), 159.8 (C-3, 5), 141.3 (C-1), 131.6 (C-1'), 129.2 (C-α), 128.8 (C-2', 6'), 127.9 (C-β), 115.2 (C-3', 5'), 105.9 (C-2, 6) 102.9 (C-4), 55.8 (4'-OCH3) (9).
In-vitro monoamine oxidase inhibition assay
Preparation of rat liver homogenates
MAO was partially purified by isolation of mitochondria from rat liver homogenates by a slightly modified with Holt’s method (13). Briefly, male Wistar rats (280–300 g) were euthanised by cervical dislocation and livers dissected out, washed in ice-cold sodium phosphate buffer (0.2 M, pH 7.6), Liver tissue was homogenized 1:10 (w/v) in 0.3 M sucrose. The homogenate was centrifugated at 1000 ×g for 10 min, the supernatant was draw off, the pellet was centrifugated at 1200 ×g for 15 min after washed with 20 mL 0.3 M sucrose. Supernatants were combined and further centrifuged at 10,000 ×g for 30 min to obtain mitochondrial pellet. The pellet was resuspended in 4 mL of phosphate buffer (0.2 M; pH 7.6) and stored at 4 °C. Suspensions of MAO were diluted eight times before use, and it must be used up in 7 days.
monoamine oxidase assay
Monoamine oxidase inhibition activity was measured in the 96-well microplates according to the method reported by Holt et al. with some modifications (Holt et al., 1997). Briefly, 40 μL enzyme and 40 μL sample solution were placed in 96-well microplates and pre-incubated at 37 °C for 20 min. The reaction was started by adding 120 μL amino substrate (2.5 mM tyramine in sodium phosphate buffer), 40 μL chromogenic solution (1 mM vanillic acid, 0.5 mM 4-aminoantipyrine, 4 U/mL peroxidase in sodium phosphate buffer), and the total solution was incubated at 37 °C for 60 min. Optical densities were measured at 490 nm. Blanks were set up by adding 40 µL buffer solutions instead of 40 µL sample solution. Blank negative controls were set up by adding 160 µL buffer solutions instead of 40 µL sample solution and 120 µL substrate solution. Sample controls were set up by adding 120 µL buffer solution instead of 120 µL substrate solution aiming to deduct sample background. The inhibition rate (%) was calculated by the following equation:
Inhibition rate% = × 100%
Inhibitory activity was expressed as the mean of 50% inhibitory concentration (IC50), obtained by interpolation of concentration-inhibition curves. The selectivity of sample against MAO-A and MAO-B were also evaluated in this assay. To test specific MAO-A activity, the rat liver homogenate was pre-incubated (37 °C; 30 min) with 500 nM pargyline (a selective inhibitor of MAO-B) to entirely inhibit MAO-B. To test specific MAO-B activity, the rat liver homogenate was pre-incubated (37 °C; 30 min) with 500 nM clorgyline (a selective inhibitor of MAO-A) to entirely inhibit MAO-A. After the enzyme was pre-incubated, the IC50 values of sample against MAO-A and MAO-B were determined respectively according above method.
Molecular modeling
In order to further study of structure activity relationship, we have docked three representative compounds 2, 4, 8 into the active site of MAO. The docking calculation was performed in Schrödinger program. The representative crystal structures of MAO-A with harmine (PDB code: 2Z5X) and MAO-B with safinamide (PDB code: 2V5Z) were obtained from the Protein Data Bank. Protein Preparation Wizard Panel tool was used to prepare the protein which include removing crystallographic water molecules, adding hydrogen atoms, assigning partial charges with the OPLS-2005 force field, assigning protonation states and minimizing the structures. Three dimensional structures of compounds 2, 4, 8 were built in ChemBio3D Ultra 11.0 and the geometry was optimized with MM2 force field. Then the Ligprep module (LigPrep, version 2.5, Schrödinger, LLC, New York, NY, 2012) was used to assign protonation states at a target pH value of 7.0 ± 2.0. Docking grid boxes of MAO-A and MAO-B were defined by centering on the ligand in 2Z5X and 2V5Z respectively. The molecular docking was performed using the Glide (Glide, version 5.8, Schrödinger, LLC, New York, NY, 2012) in standard precision (SP) mode. As a result, highest scoring docking poses of compound 2, 4, 8 in two kinds of protein receptors were selected for further analysis.
Results and Discussion
Repeated column chromatography resulted in the isolation of eight compounds 1-8 (Figure 1). By comparing NMR data with those published in the literature, 1-8 were identified as piceatannol 1, resveratrol 2, piceid 3, rhapontigenin 4, piceatannol-3'-O-β-D-glucopyranoside 5, rhapontigenin 6, catechin 7 and desoxyrhapontigenin 8. The inhibitory effects of the isolated compounds 1-8 on MAO, were showed in Table 1. As shown in Table 1. compounds 2, 3, 6 and 7 exhibited weak inhibitory activity against mixed type MAO; compounds 4, 5 exhibited moderate inhibitory activity; whereas, compounds 1 and 8 displayed significant inhibitory activity with the IC50 values 16.4 and 11.5 µM, respectively, which were close to those of positive control ((+) Iproniazid phosphate IC50 = 7.0 µM). The selectivity of compounds 1-8 against MAO-A and MAO-B were also evaluated. The results (Table 1.) showed that compounds 4˴6˴8 preferred to inhibit MAO-A rather than MAO-B with selectivity values ([IC50 of MAO-B]/ [IC50 of MAO-A]) of 4.74, 10.01 and 9.42 respectively. The preliminary structure-activity relationships (SARs) could be drawn from the data of Table 1. as follows: (1) The activity of stilbenes (compounds 1-6, 8) was better than that of flavanol (compound 7). (2) The methoxyl at the C-4’ position of stilbenes plays an important role for MAO-A selectivity. For example, compounds 4, 6, 8 which all have a methoxyl at the C-4’ position exhibited relatively strong inhibition potencies towards MAO-A compared to MAO-B. (3) Only one methoxyl group at C-4’ position and no other substituent groups on B-ring, were favorable for MAO (mixed-type MAO, MAO-A and MAO-B) inhibition. For example, compound 8, with only one methoxyl group at C-4’ position showed the best inhibitory activity, and the activity of compound 2 (a demethylated product of 8) decreased almost 8 times for mixed type MAO, 20 times for MAO-A, 4 times for MAO-A, compared to that of 8. This influence was also reflected between 4 and 8, 4 with an extra hydroxyl at C-3’ position having weaker activity than 8. The fact that compounds 1, 3, 5 with no methoxyl group at C-4’ and compound 6 with methoxyl group at C-4’ but also with an extra groups at C-3’, showed weaker activity than 8, further supported the conclusion that only one methoxyl at C-4’ position were important for MAO inhibition. (4) The influences of the glycosyls at A-ring on activity have no rules. For example, the activity of 6 (the glucoside of 4) against mixed-type MAO and MAO-B have a little decrease compared to 4, but the activity of 3 (the glucoside of 2) against MAO-A and MAO-B have a little increase compared to 2.

The structures of compound 1-8.
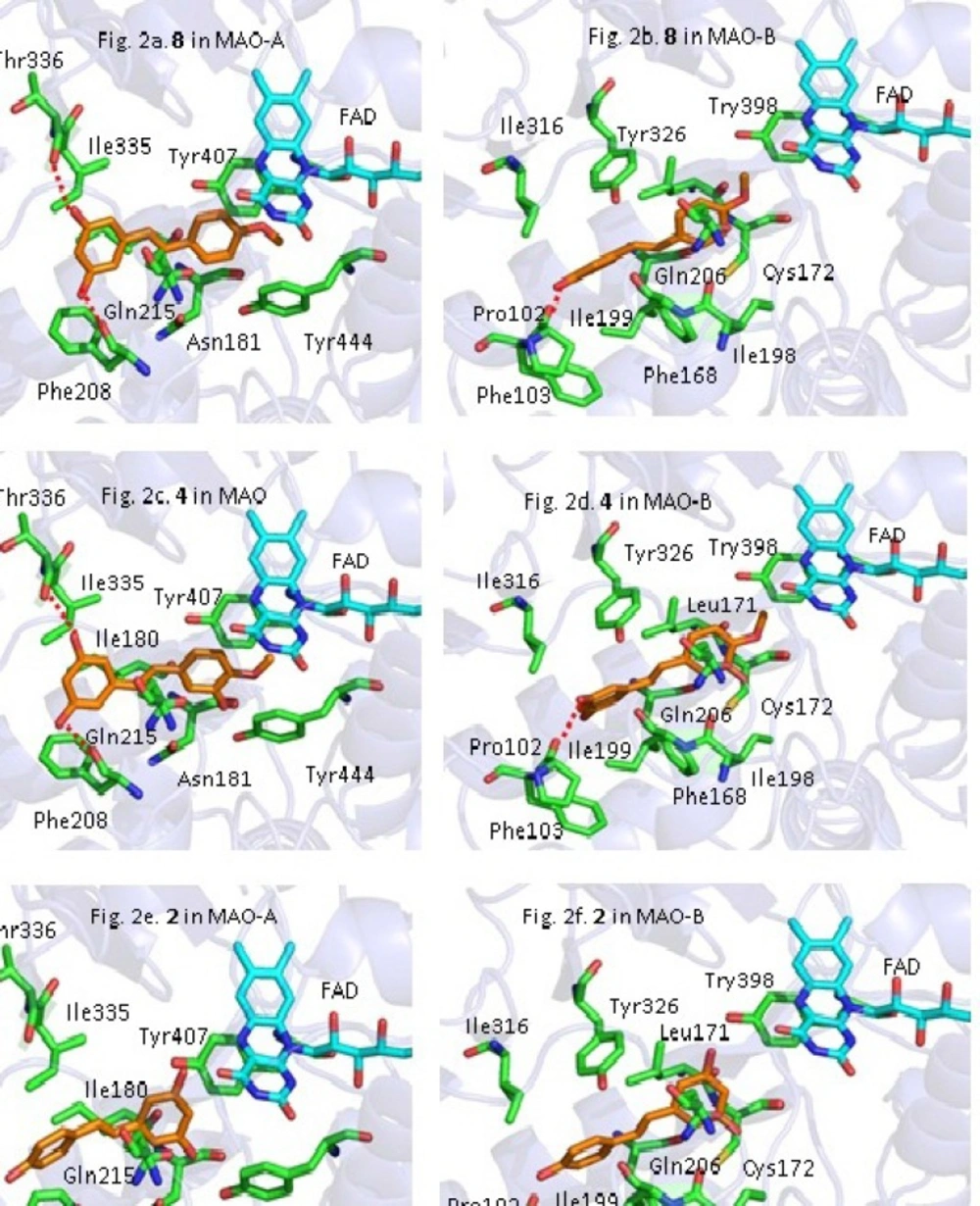
The predicted binding mode of compounds 2, 4, 8 to MAO-A and MAO-B. The MAO-A and MAO-B was shown as ribbon model. The side chains of the active site residues (green or blue sticks) and compounds 2, 4, 8 (magenta sticks) were represented as stick model
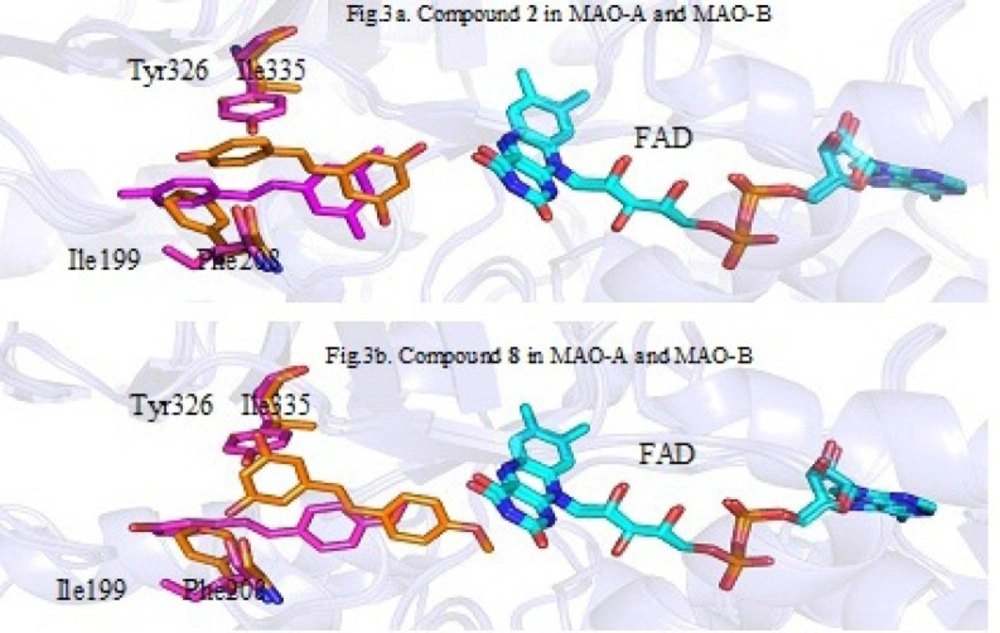
The superimposition of the ligand in MAO-A (orange) and MAO-B (magentas).
| sample | IC50 (Mixed type MAO, µM)a | IC50 (MAO-A, µM)a | IC50 (MAO-B, µM)a | Selectivity Values |
|---|---|---|---|---|
| Piceatannol (1) | 16.4±1.5 | 18.7±1.5 | 18.7±1.9 | 1 |
| Resveratrol (2) | 88.2±3.2 | 194.3±4.8 | 150.2±5.7 | 0.77 |
| Piceid (3) | 85.0±3.8 | 87.1±2.1 | 96.3±3.4 | 1.10 |
| Rhapontigenin (4) | 38.1±2.1 | 27.0±1.6 | 128.1±4.8 | 4.74 |
| Piceatannol-3'-O-β-D- glucopyranoside (5) | 47.3±3.2 | 34.0±2.8 | 43.2±2.4 | 1.27 |
| Rhapontigenin (6) | 70.4±.3.8 | 26.0±1.2 | 260.3±5.6 | 10.01 |
| Catechin (7) | 140.5±5.4 | 130.0±4.3 | 125.6±4.2 | 0.97 |
| Desoxyrhapontigenin (8) | 11.5±.1.1 | 4.5±1.1 | 42.4±2.7 | 9.42 |
| (+)Iproniazid Phosphateb | 7.0±0.6 | 6.1±0.3 | 9.0±0.5 | 1.47 |
In-vitro quantitative inhibition of MAO by 1–8.
| Compound 8 | Compound 4 | Compound 2 | |
|---|---|---|---|
| MAO-A(Docking/(Kcal/mol) ) | -10.06 | -9.25 | -8.36 |
| MAO-A (test, IC50/µM)) | 4.5 | 27 | 194 |
| MAO-B(docking/(Kcal/mol) ) | -9.24 | -8.95 | -8.62 |
| MAO-B (test, IC50/µM)) | 42 | 128 | 150 |
The calculated binding free energy of 2, 4, 8with MAO.
Compound 2, 4, 8 were selected to explore the possible interaction mode between ligand and the active site of MAO-A and MAO-B. Figure 2. showed the interaction mode of 2, 4, 8 with MAO-A and MAO-B.
Among the three compounds, 8 had the best binding affinity with both MAO-A and MAO-B. As shown in Figure 2a and 2b. the two hydroxyl on the ring A of 8 formed hydrogen bonds with Thr336 and Phe208 residues of MAO-A, respectively (Figure 2a). The methoxyl on the ring B of 8 had a strong Van der Waals interaction with FAD in MAO-A (Figure 2a). For the interaction mode between 8 and MAO-B, one hydrogen bond formed by the hydroxyl group of the ring A with Pro102 was observed, and the amino acid residues Ile199˴Leu71˴Tyr326 and Phe168 showed relatively strong hydrophobic interaction with compound 8.
Compound 4 adopted the same conformation as 8, as seen in Figure 2c. 2d. However, the affinity of 4 with MAO-A and MAO-B were weaker than those of 8. In terms of MAO-A, the hydroxyl at C-3’ position of 4 produce strong steric repulsion with Tyr444 which caused some degree of twist of the ring B and also reduced the Van der Waals interaction between the 4’-OCH3 and FAD. For MAO-B, the ring A of 4 had some degree of twist which led to π-π stacking interaction of Phe168 and Phe103 with ligand decrease. Above results indicated the introduction of -OH in ortho position of -OCH3 would result in decrease of interaction of active pocket of enzyme and ligand.
Figure 2e and 2f. showed the interaction mode of 2 with MAO-A and MAO-B. As demonstrated in Figure 2e and 2f. the conformation of 2 are drastically changed, compared with 8 and 4. The location of ring A and ring B of compound 2 caused the loss of hydrogen bond interaction with specific residues and decrease of the van der Waals interaction with FAD. As a result, the activity of 2 against MAO-A and MAO-B decreased remarkably. The docking results above, revealed that the removal of -OCH3 at C-4’ was disadvantageous to activity.
To explain the reason that the methoxyl at the C-4’ position of stilbenes plays an important role for MAO-A selectivity, We aligned the conformations of compound 2 and 8 in MAO-A to those in MAO-B. Figure 3. is the superimposition of the location of compound 2 and 8 in MAO-A and MAO-B binding pocket (orange is for MAO-A and magentas is for MAO-B). Although the structures of MAO-A and MAO-B have some similarity, there are still some key amino acids different in binding pocket (MAO-A: Phe208, Ile335; MAO-B: Ile199, Tyr326) which cause ligands are closer to FAD in MAO-A than in MAO-B when binding to the active site. Since compound 2 with no -OCH3 at C-4’ only has relatively weak van der Waals interaction with FAD in MAO-A or MAO-B, the distance of 2 and FAD less affected the MAO inhibitory activity. Thus, the selectivity of 2 against MAO-A and MAO-B is weak. However, for compound 8, due to the presence of a -OCH3 at C-4’, the van der Waals interactions between -OCH3 and FAD tend to play a key role in the interaction of ligand and active pocket. Because the distance between 8 and the FAD in MAO-A pocket is shorter than that in MAO-B, the hydrophobic interactions of 8 with MAO-A will be stronger than that of 8 with MAO-B. This explain the reason that 8 prefer to inhibit MAO-A rather than MAO-B.
In addition, the binding free energy of 2, 4 and 8 were also calculated which were found to correlate well with experimental results, as shown in Table 2.
Up to now, many pant extracts have been reported with potential MAO inhibitory activity which suggested that plant is a good source for finding new MAOIs. For example, in 2013, Mazzio, et al. (14) screened 905 natural extracts for their human MAO-B inhibitory activity. The data showed some extracts showed strong inhibitory activity and few of that inhibited MAO-B within therapeutic range. They also isolated some compounds from these herbs. Our study further conformed the plant is a good resource of natural MAOIs
Conclusion
In conclusion, seven MAO inhibitors with stilbene skeleton (compound 1-6, 8) have been obtained and their preliminary structure–activity relationships (SARs) were also discussed. The result shows that: (1) The 4’-OCH3 of stilbenes were important for MAO inhibitory activity and selectivity. (2) Except for 4’-OCH3, the presences of other substituent group at ring B were disadvantageous to MAO inhibition.
Conflicts of interest
The authors declare that there are no conflicts of interest.
