
Introduction
Mercury is a heavy metal that has known as a toxicant in living organisms and exposure to mercury compounds continue to be prevalent and significant health risks to human (1). Approximately one million tons of metallic Hg has been extracted from ores during the past five centuries (2). Original routes of human exposure to Hg are environmental contaminant, industrial usage and health care. Multiple sclerosis is a chronic and progressive autoimmune disease in the central nervous system (CNS) of young adults, with over 2.5 million individuals affected worldwide (3). MS has almost unknown etiology and no effective cure at the time being. it`s treatment is supportive and symptomatic (4). Mitochondria extensive dysfunction and concomitant oxidative stress can cause many neurodegenerative diseases such as multiple sclerosis (5). There are many study that show relation between mercury exposure and MS for example mercury-containing amalgam may exert an important risk for patients with autoimmune diseases (6). Mercury concentrations in the cerebrospinal fluid and blood serum of MS patients is higher than healthy individuals (7) .It has already been demonstrated that inhibition of myelin production can be resulted from toxic effect of mercury on human oligodendrocytes (8).
In this study for evaluation of relationship progression of MS and repeated exposure of mercury we used EAE model to induce MS in C57BL/6 mice, then neurobehavioral symptoms observed and recorded. Finally We sacrificed mice at day 28 following EAE induction and then brain mitochondria were isolated to examine mitochondrial damage parameters such as mitochondrial swelling, ROS formation, collapse of MMP and cytochrome c release.
Material and Methods
Animals
All experiments were performed on female C57BL/6 mice (Razi Institute, Karaj, Iran) weighing 20-25 g (12 weeks).The animals were kept under a 12 h light/dark cycle in a temperature controlled (22. ± 2 °C) environment with free access to food and water ad libitum. All studies and animal care procedures were accomplished in accordance with the local ethics committee for animal experimentation and in compliance with guidelines of Shahid Beheshti University of Medical Sciences.
Induction of EAE and experimental design
EAE model for MS was induced in mice with MOG35–55 peptide emulsified in complete Freund’s adjuvant (CFA) using Hooke kits (Hooke Laboratories, Lawrence, MA, USA) according to the manufacturer’s instruction. All drugs and their respective vehicles were injected intraperitoneally. The intact (no EAE) group received CFA and pertussis toxin without MOG. Mercury sulfides suspended in carboxymethyl cellulose (CMC) then it exposed to mice oral daily (1 mg/Kg/day) for 7 days from the onset of EAE induction. (9).
The following groups were used in this study: control group received CFA and pertussis toxin without MOG, EAE group, mercury sulfide group, and EAE + mercury sulfide group. The number of animals in each group were 8(n=8).
Neurological assessment
Mice were daily observed to record behavioral and neurological signs until day 28 after immunization. The signs of EAE were scored protocol of Stromnes et al. (10).
Mitochondrial preparation
At 28th day after induction EAE mice were sacrificed and brain and spinal cord mitochondria were isolated using differential centrifugation (11). Mitochondria were prepared freshly for each experiment and used within 4 h of isolation.
Determination of mitochondrial ROS level
The mitochondrial ROS measurement was performed using the fluorescent probe DCFH-DA. The fluorescence intensity of DCF was measured using Shimadzu RF-5000U fluorescence spectrophotometer at an excitation wavelength of 488 nm and emission wavelength of 527 nm (12, 13).
Determination of the MMP
Mitochondrial uptake of the cationic fluorescent dye, rhodamine123, has been used for the estimation of mitochondrial membrane potential. The mitochondrial fractions (0.5 mg protein/mL) were incubated with 10 µM of rhodamine 123 in MMP assay buffer. The fluorescence was monitored using Shimadzu RF-5000U fluorescence spectrophotometer at the excitation and emission wavelength of 490 nm and 535 nm, respectively (14, 15) .
Determination of mitochondrial swelling
Briefly, isolated mitochondria were suspended in swelling buffer (70 mM sucrose, 230 mM mannitol, 3 mM HEPES, 2 mM tris phosphate, 5 mM succinate and 1 µM of rotenone). The absorbance was measured at 549 nm at 15 min time intervals with an ELISA reader (Tecan, Rainbow Thermo and Austria) (13, 16).
Cytochrome c release assay
The concentration of cytochrome c was determined through using the Quantikine Rat/Mouse Cytochrome c Immunoassay kit provided by R&D Systems, Inc. (Minneapolis, Minn.) according to manufacturer’s instruction (15).
Statistical analysis
The results are expressed as mean ± SEM and were analyzed using Graph Pad
Prism (version 5, Graph pad Software Inc.). A one-way or two-way analysis of variance (ANOVA) followed by Bonferroni’s tests were used for multiple comparison. For all statistical analyses, p<0.05 was considered significant.
Results
EAE scoring
As shown in Figure 1 behavioral and neurological signs were increased in EAE induced group which repeatedly received Hg. In the Hg2+treated group, there were also some behavioral and neurological signs.
Measurement of ROS generation
As shown in Figure 2. ROS significantly (P<0.05) increased in the brain mitochondria isolated from (EAE + Hg2+) group compared with EAE group. ROS were also significantly (P<0.05) increased in EAE group compared with control group.
Determination of mitochondrial membrane potential
As shown in Figure 3. MMP significantly decreased in the brain mitochondria isolated from EAE + Hg group compared with EAE group.
Measurement of mitochondrial swelling
As shown in Figure 4. collapse of optical absorbance at 540 nm which is consistent with mitochondrial swelling, was assayed. Our results showed that of repeated administration of mercuric sulfide significantly enhanced swelling in the brain mitochondria isolated from (EAE + Hg).
Cytochrome c release
As shown in Figure 5. there is significant enhancement in the release of cytochrome c from brain mitochondria in the (EAE + Hg) group compared with EAE group.
Discussion
Using EAE scoring test we proved that, mercury causes behavioral, neuromuscular, sensorimotor disturbances in EAE + Hg treated mice compared to Hg group (Figure 1). Our results showed that in Hg accelerated behavioral dysfunction in EAE model. Moreover there are many previous studies that indicate relationship between MS and mercury. Mercury is a heavy metal that exerts acute and chronic toxic effects on the human body including the nervous and immune systems (17). One of the main pathways of mercury toxicity is damage to mitochondrial function (18). Malfunction of mitochondria has been associated to neurodegenerative diseases (19). The functionality of mitochondrial preparations is very useful in the study of the processes underlying pathologies of these diseases. It has already proven that use of isolated mitochondria is a very powerful tool in the study of pathologies of many neurodegenerative (20). Results of ROS measurement in isolated mitochondria showed that ROS formation was significantly increased in EAE + Hg+2 group (Figure 2). Following the rise of reactive oxygen species formation and consecutive oxidative damage to mitochondria, mitochondrial functions are impaired. Our findings regarding mitochondrial membrane potential collapse (Figure 3.) and mitochondrial swelling (Figure 4.) in EAE + Hg group proved this hypothesis. Previous studies with mercury have also shown increased damage through ROS formation. Our results suggest in this animal model, substantial ROS produced by mercury increases and accelerates neural damage compared to EAE group. The increased mitochondrial ROS formation can cause oxidation of a lipid membrane which consequently results in disruption of the mitochondrial membrane and subsequently the collapse of mitochondrial membrane potential (MMP) and cytochrome c release. (21). A number of studies have reported mitochondrial defects in MS. We also showed that in EAE model mitochondrial damages increased which with repeated exposure with mercury these damage significantly increased.
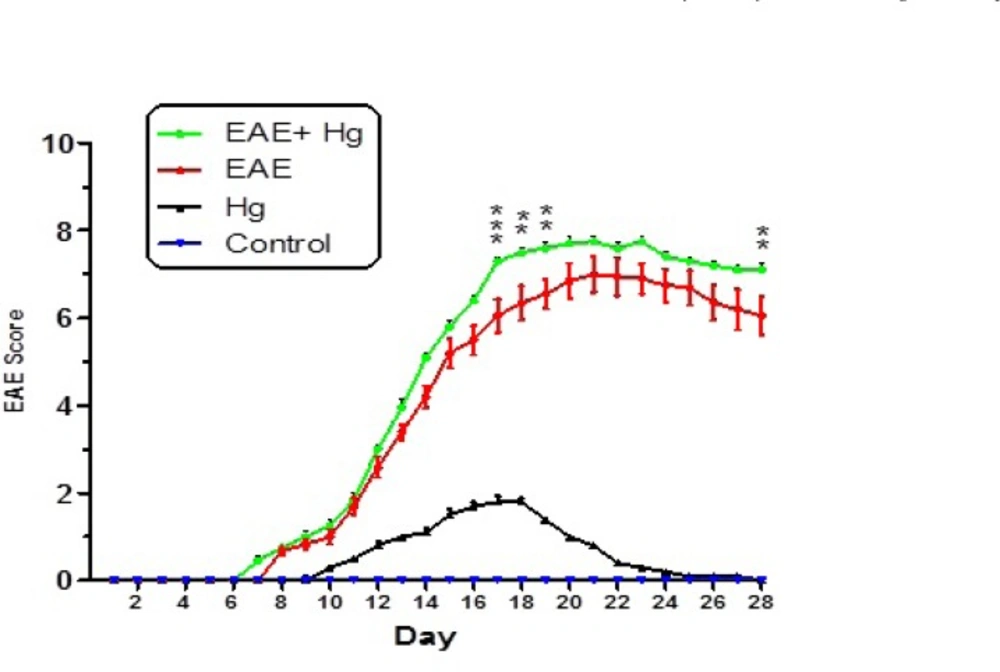
Behavioral and neurological signs in mice experimental groups. Values were presented as score of signs as described in materials and methods (n = 8). ** represents P < 0.01 significant difference, *** represents P < 0.001 significant difference, compared to EAE induced mice group.
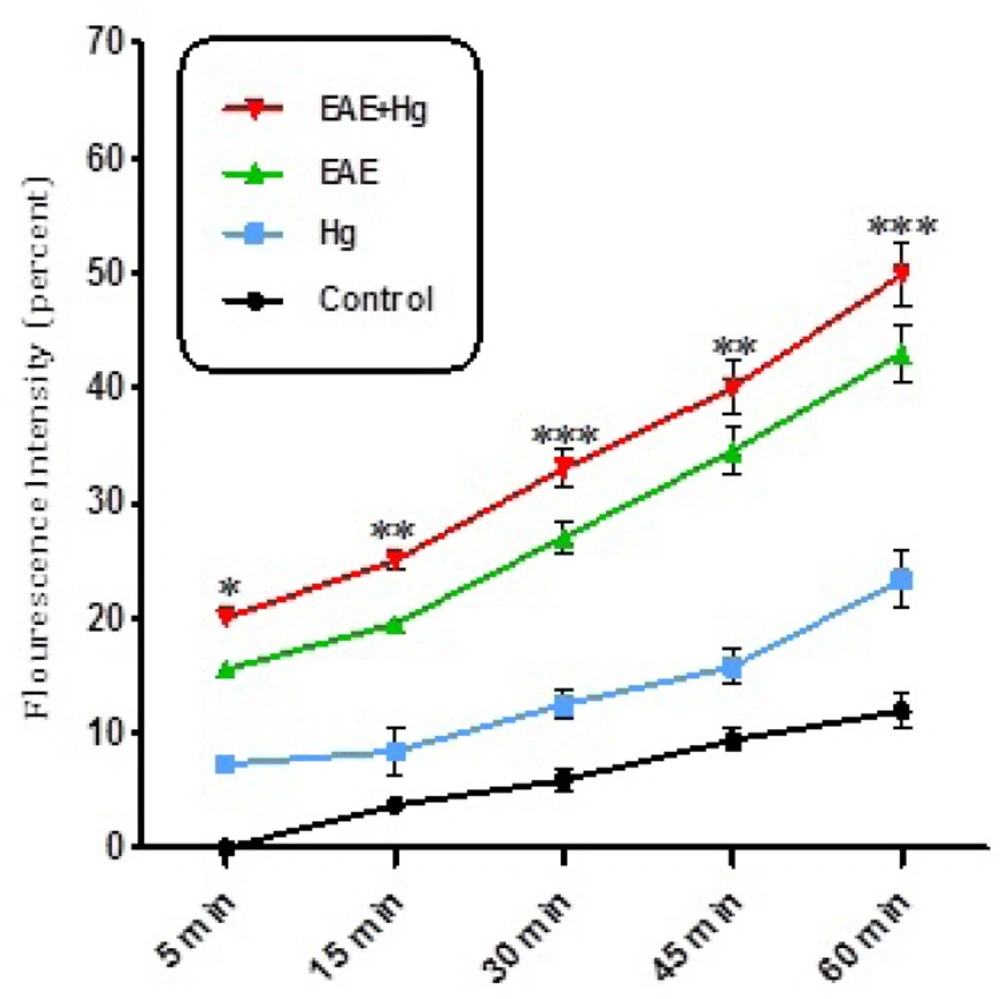
ROS formation. Exposure to mercury (Hg) intensified ROS formation in EAE + Hg group compared to EAE group. Values have been presented as percent ROS formation (n = 8). *** represents significant difference between control and EAE groups, also $$$ represents significant difference (P < 0.001) between EAE and EAE+Hg2+ groups
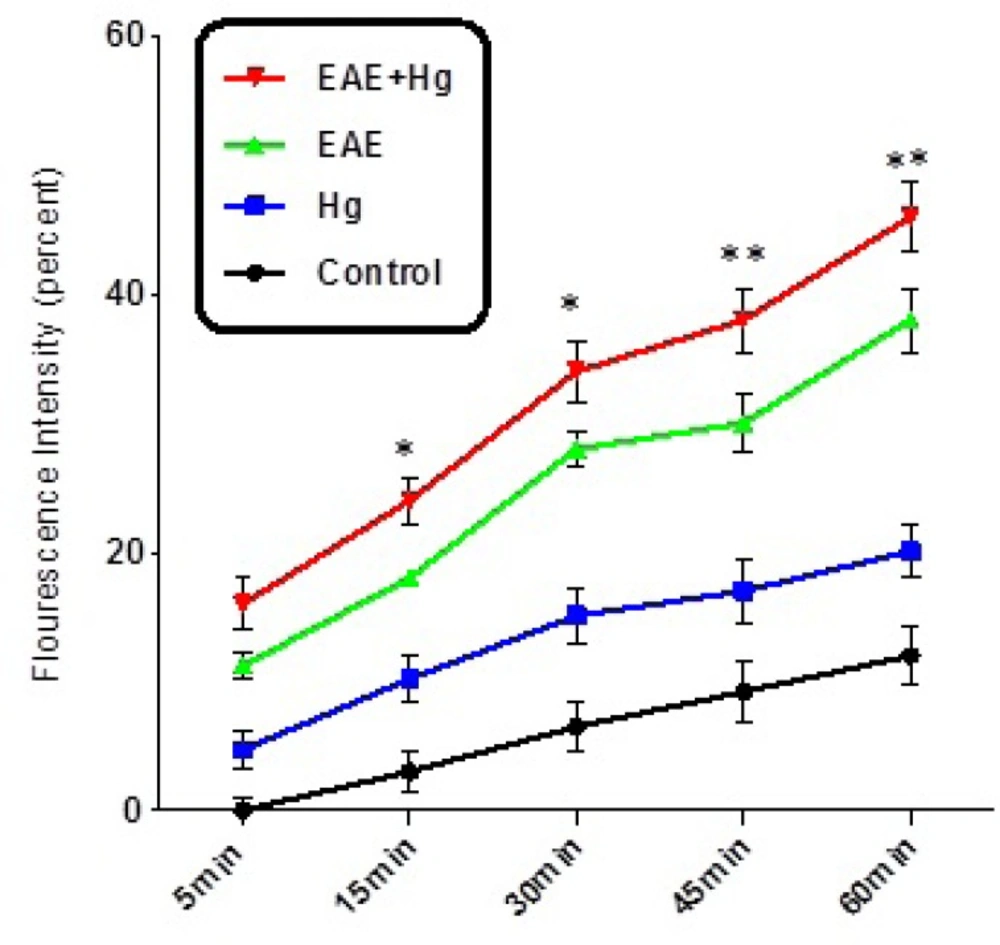
Decline of MMP. Decline of MMP significantly decreased in EAE + Hg group compared to EAE group. Values expressed as percent of MMP collapse (n = 8). *** represents significant difference between control and EAE groups, also $$$ represents significant difference (P < 0.001) between EAE and EAE + Hg groups
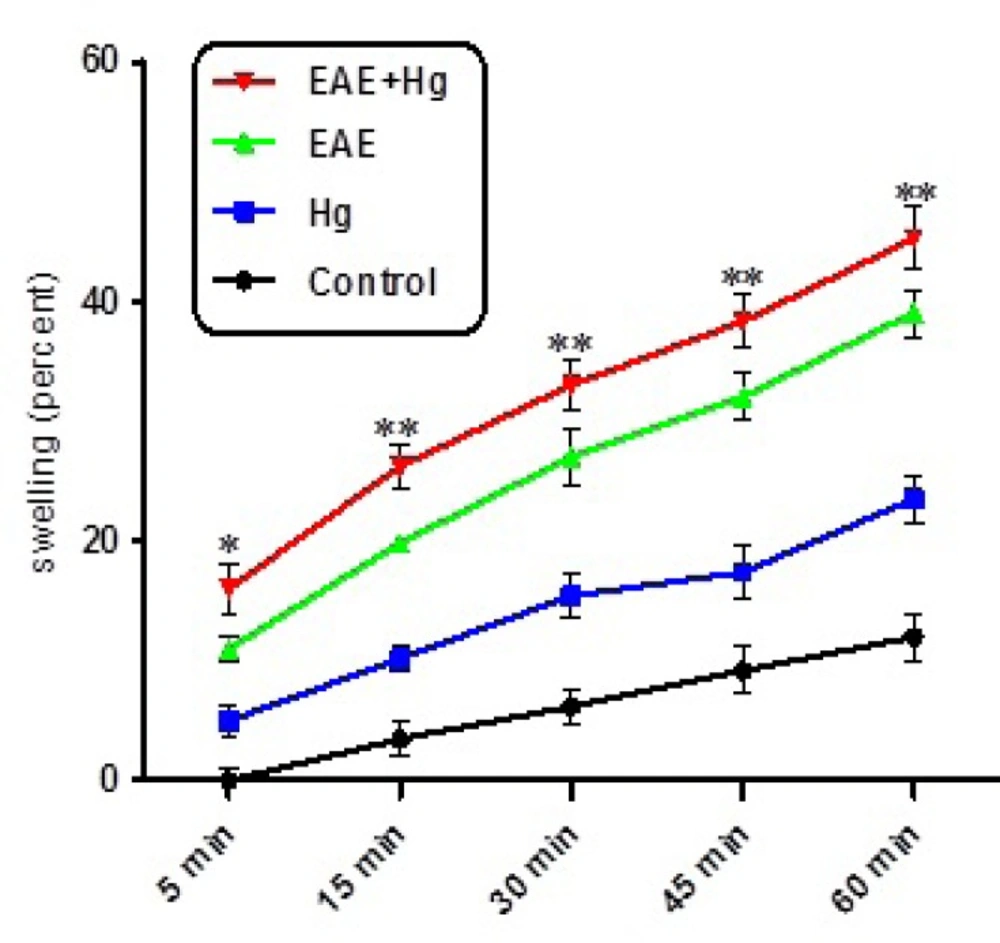
Mitochondrial swelling. Hg enhanced mitochondrial swelling in EAE + Hg group. Values have been presented as decreasing absorbance at 450 nm (n = 8). *** represents significant difference between control and EAE groups, also $$$ represents significant difference (P < 0.001) between EAE and EAE + Hg groups
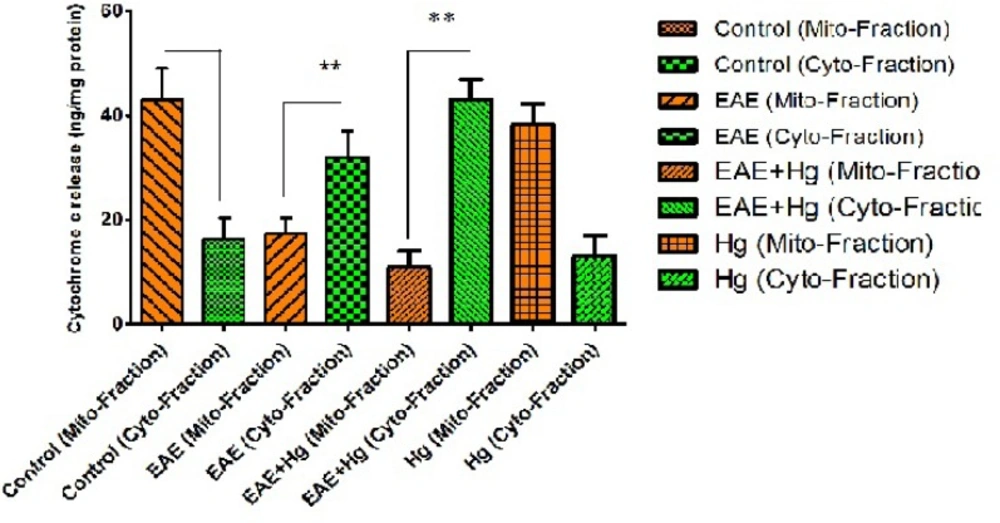
Cytochrome c release. In EAE + Hg and EAE groups cytochrome c released. Values have been presented as mean ± SD (n = 8). *** represents significant difference (P < 0. 001) compared to EAE induced group. Also $$$ represents significant difference (P < 0. 001) between EAE + Hg and EAE groups
Our results provided evidence that cumulative mitochondrial damages result in mercury accelerates progression MS through mitochondrial ROS formation that lead to collapse MMP, mitochondrial swelling and finally initiation of apoptosis signaling through mitochondrial pathway. To our knowledge this the first report that provides relationship between mitochondrial events and progression of MS through apoptosis by mercury in brain.
