General experimental procedures
A Thermo pH-meter, a BioTek Power Wave XS, an Elma S15 ultrasonic bath and a vortex (LMS Co. LTD) were used for the activity assays. Ethanol, hexane, diethyl ether, chloroform, toluene, dichloromethane, methanol, potassium acetate, sulphuric acid, aluminium nitrate nonahydrate, aluminium chloride, ABTS, sodium acetate, nutrient broth, boric acid, nutrient agar, butylated hydroxytoluene were purchased from Merck (Germany), DPPH, -carotene, H2O2, quercetin, pyrocathecol, acetic acid, sodium methoxide, Tween 40, DTNB, copper (II) chloride dihydrate (CuCl2.2H2O), linoleic acid, neocuproine, EDTA, acetylcholinesterase, butyrylcholinesterase from Sigma (Germany), -tocopherol, acetylthiocholine iodide from Aldrich (Germany), galanthamine hydrobromide from Sigma-Aldrich (Germany), BHT from Fluka (Germany), sterile blank disc and antbiotic disc from Oxoid (United Kingdom), petroleum ether, sodium dihydrogen phosphate, sodium carbonate, sodium hydrogen phosphate, ammonium acetate from Reidel de Haen (Germany).
Plant material
A whole plant of Trifolium angustifolium L. var. angustifolium L. was collected from western Turkey (Istanbul)) in April 2012 by Dr. Abdulselam Ertaş and identified by Dr. Mine Koçyiğit (Istanbul University, Faculty of Pharmacy, Dept of Pharmaceutical Botany). This specimen has been stored at the Herbarium of Istanbul University (ISTE 98260).
Preparation and GC/MS conditions for essential oil
Essential oil was obtained using a Clevenger apparatus from the whole parts of plant, which was crumbled into small pieces and soaked in distilled water for 3 h. The obtained essential oil was dried over anhydrous Na2SO4 and stored at +4 °C for a sufficient period of time. The essential oil was diluted using CH2Cl2 (1:3 volume/volume) prior to GC/FID and GC/MS analysis. GC/FID performed using Thermo Electron Trace GC FID detector and GC/MS performed using same GC and Thermo Electron DSQ quadrupole for MS.
The GC oven temperature was kept at 60 °C for 10 min and programmed to 280 °C at a rate of 4 °C/min and then kept constant at 280 °C for 10 min. A nonpolar Phenomenex DB5 fused silica column (30 m 0.32 mm, 0.25 μm film thickness) was used with helium at 1 mL/min (20 psi) as a carrier gas. The split ratio was adjusted to 1:50, the injection volume was 0.1 μL, and EI/MS was recorded at 70 eV ionization energy. The mass range was
m/z 35–500 amu. Alkanes (C8-C24) were used as reference points in the calculation of Kovats Indices (KI) by the same conditions (
12,
13).
Identification of the compounds was based on comparing their retention times and mass spectra with those obtained from authentic samples and/or the NIST and Wiley spectra as well as data from the published literature. GC/FID and GC/MS were replicated three times. (Mean RSD % < 0.1).
Esterification of total fatty acids with GC/MS conditions
A hundred milligram of the petroleum ether extract was refluxed in 0.1 M NaOH solution in 2 mL of methanol during 1 h, the solution was cooled and 5 mL of water was added. The aqueous mixture was neutralized with 0.5 mL of HCl solution, it was extracted with diethyl ether: hexane (3.5: 1: 1 mL). The separating organic phase was washed with 10 mL water, and dried over anhydrous Na
2SO
4. The solvent was evaporated in vacuum and then fatty acid methyl esters were obtained (
14). The analyses was performed using a Thermo Scientific Polaris Q GC-MS/MS. GC/MS procedure described by Sabudak et al. was applied (
14).
Preparation of plant extracts
Whole plants of T. angustifolium var. angustifolium (100 g) were dried, powdered, and then sequentially macerated with petroleum ether, acetone, methanol, and water for 24 h at 25 °C. After filtration, the solvents were evaporated to obtain crude extracts. This yielded 0.67% petroleum ether extract, 0.70% acetone extract, 4.5% methanol extract, and 2.8% water extract (w/w).
Determination of total phenolic and flavonoid contents of extracts
The amounts of phenolic and flavonoid contents in the crude extracts were expressed as pyrocatechol and quercetin equivalents, and they were calculated according to the following equations (
15,
16):
Absorbance = 0.0128 pyrocatechol (μg) + 0.0324 (R2 = 0.9920)
Absorbance = 0.1701 quercetin (μg) – 0.7078 (R2 = 0.9939)
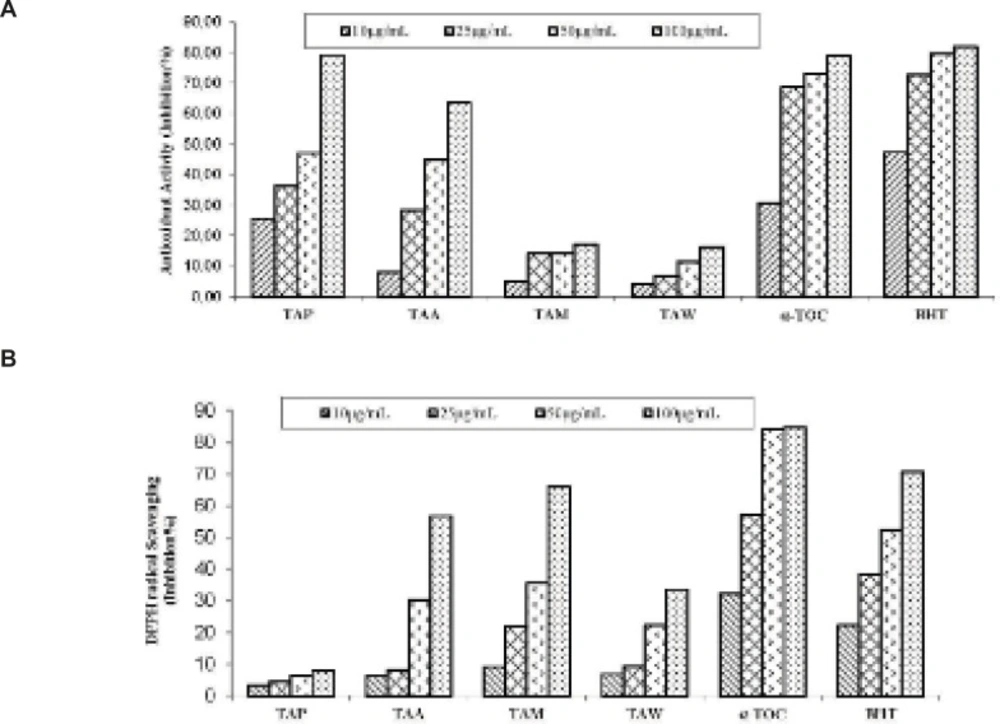
Antioxidant activityof extracts
-Carotene bleaching method
0.5 mg of -carotene in 1 mL of chloroform was added into linoleic acid (25 L) and Tween 40 emulsifier (200 mg) mixture. After evaporating chloroform, 100 mL of distilled water saturated with oxygen was added followed by shaking, 160 μL of this mixture was transferred into different test tubes containing 40 μL of the sample solutions at different concentrations. The emulsion was added to each tube, the zero time absorbances of the values were read at 470 nm. The mixture was incubated for 2 h at 50
0C (
17,
18).
Free radical scavenging activity method
0.1 mM, 160 µL of DPPH solution in methanol was added to 40 µL of sample solutions in methanol at different concentrations. After 30 min. the absorbance values were read at 517 nm. The DPPH free radical scavenging potential was calculated using the following equation (
18,
19):
AControl is the initial concentration of the DPPH•
ASample is the absorbance of the remaining concentration of DPPH• in the presence of the extracts or positive controls.
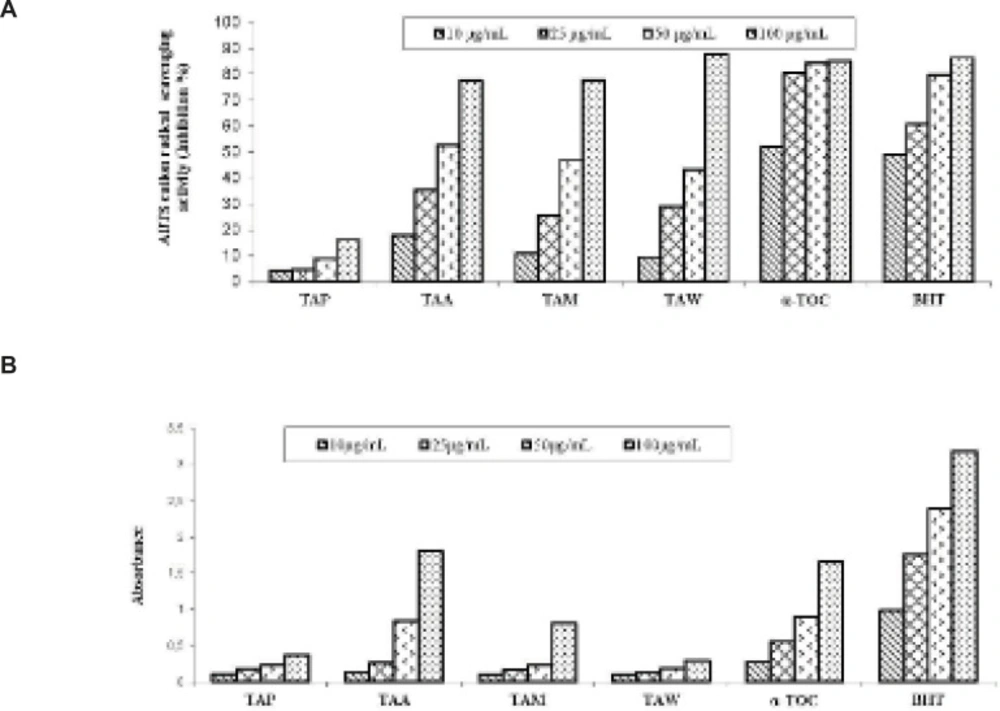
ABTScation radical decolorization assay
Seven milimolar ABTS in H
2O was added to 2.45 mM potassium persulfate to produce ABTS
•+ and solution was stored in the dark at 25 ºC for 12 h. The prepared solution was diluted with ethanol to get an absorbance of 0.700 ± 0.025 at 734 nm. ABTS
•+ solution (160 µL) was added to each sample solution at different concentrations. After 30 min, the percentage inhibition at 734 nm was read for each concentration relative to a blank absorbance (methanol). The following equation was used to calculate the scavenging capability of ABTS
•+ (
20):
Cupric reducing antioxidant capacity (CUPRAC) method
The petroleum ether and acetone extracts were dissolved in methanol, and methanol and water extracts in distilled water to prepare their stock solution at 1000 μg/mL concentration. Aliquots of 61 mL of 1.0 × 10
−2 M copper (II) chloride, 61 μL of NH
4OAc buffer (1 M, pH 7.0), and 61 μL of 7.5 × 10
−3 M neocuproine solution were mixed,
x μL sample solution (2.5, 6.25, 12.5, and 25 μL) and (67 −
x) μL distilled water were added to make the final volume 250 μL. The tubes were stopped, and after 1 h, the absorbance at 450 nm was measured against a reagent blank (
21).
Anticholinesterase activity of the extracts
All samples were dissolved in ethanol to prepare their stock solution at 4000 μg/mL concentration. Aliquots of 150 µL of 100 mM sodium phosphate buffer (pH 8.0), 10 μL of sample solution and 20 μL BChE (or AChE) solution were mixed and incubated for 15 min at 25 ºC, and DTNB (10 μL) is added. The reaction was then initiated by the addition of butyrylthiocholine iodide (or acetylthiocholine iodide) (10 μL). Final concentration of the tested solutions was 200 μg/mL (
22). The hydrolysis of these substrates were monitored using a BioTek Power Wave XS at 412 nm.
Antimicrobial activity of extracts
Five different microorganisms including Gram-positive bacteria (
Streptococcus pyogenes ATCC19615 and
Staphylococcus aureus ATCC 25923), Gram-negative bacteria (
Pseudomonas aeruginosa ATCC 27853,
Escherichia coli ATCC 25922), and yeast (
Candida albicans ATCC10231) were purchased from Refik Saydam Sanitation Center (Turkey) and were used for detecting the antimicrobial activity of the samples. The disc diffusion method was employed for this purpose (
23). Imipenem and nystatin were used as positive controls for bacteria and yeast, respectively.
Statistical analysis
The results of the antioxidant and anticholinesterase activity assays are expressed as the mean ± SD of three parallel measurements. The statistical significance was estimated using a Student’s t-test, where p-values < 0.05 were considered significant.