
Introduction
The structure of synthetic cannabinoid agonists WIN 55,212-2 (WIN). Molecular formula (MF) of C27H26N2O3 and molecular weight (MW) of 426.51 (A) and tetrahydrocannabinol (THC), MF of C21H30O2, and MW of 314.46 (B) drawing by PubChem program (http://www.ncbi.nlm.nih.gov/pccompound).
Experimental
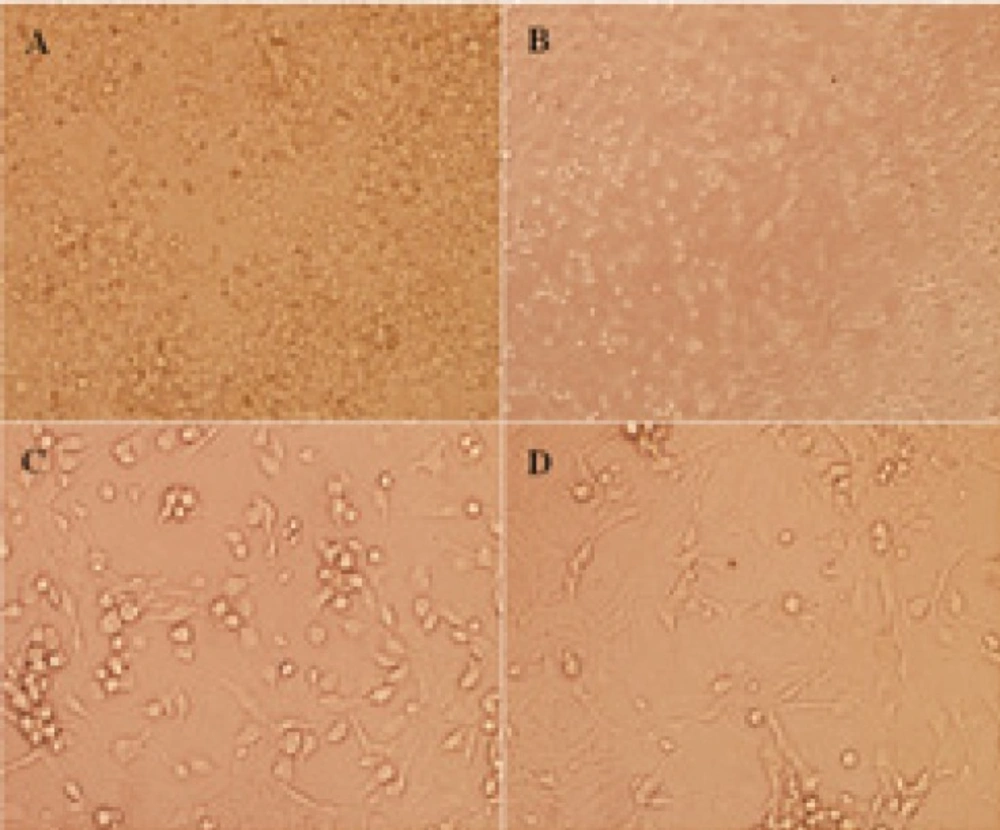
The dissociated cerebellar granular cells (CGNs) from cerebellum of postnatal 7-day-old Wistar rat pups. CGNs after 24 h seeded in 12-well culture plate were presented with × 100 magnification (A). CGNs after 24 h following cytosine arabinoside (Ara-C) added with × 100 (B) and × 400 magnification (C), respectively, using light microscope were indicated. CGNs at day 5 which all pharmacological interventions were started were presented with × 400 magnification (D).
Results
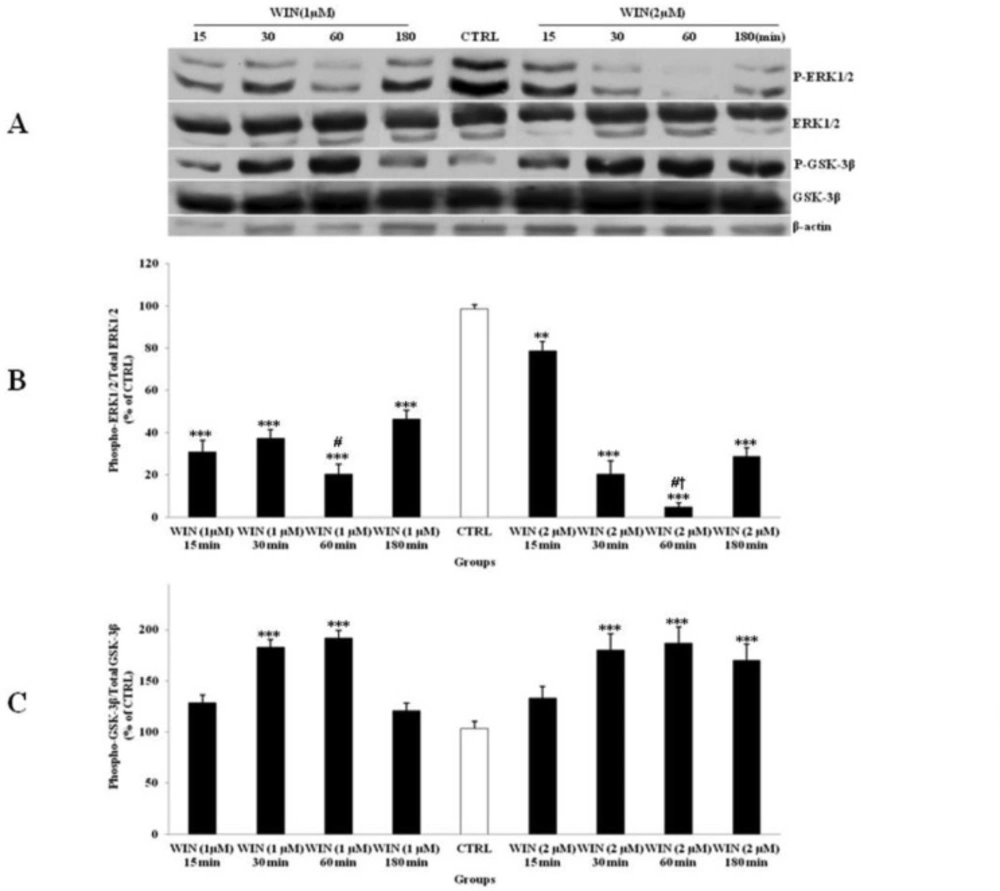
The effect of WIN 55,212-2 (WIN) as a potent cannabinoid agonist, on the phosphorylated-ERK1/2 (p-ERK1/2), total ERK1/2, phosphorylated-GSK-3β (p-GSK-3β) and total GSK-3β expression in cerebellar granular cells (CGNs). CGNs were treated with 1 and 2 µM of WIN and the cell lysates were prepared after 15, 30, 60 and 180 minutes. The protein expression was analyzed by western blotting. The p-ERK1/2 and ERK1/2 expression (A, B), and the p-GSK-3β and GSK-3β expression (A, C), were evaluated. The protein expression was normalized to β-actin as internal control (CTRL). The band intensity was measured and presented as the percent of un-treated cells (CTRL). All data were presented as Mean ± SD. Statistically significant values are presented as follows: *** p < 0.001, ** p < 0.01 and * p < 0.05, compared to CTRL; # p < 0.05, compared to respective time in two doses; † p < 0.001, compared the dose of 1 µM with the dose of 2 µM at 60 min exposure
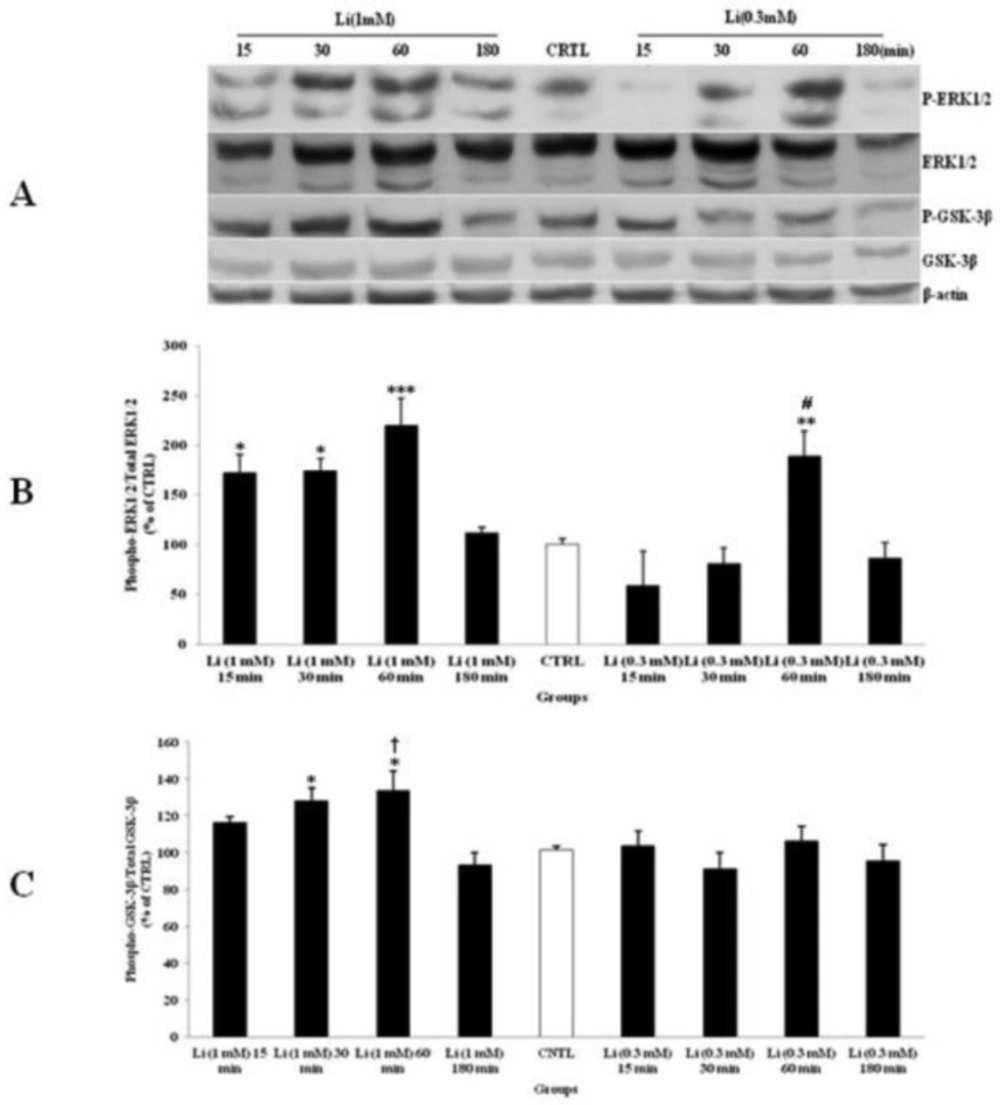
The effect of lithium (Li) as a mood stabilizer drug, on the phosphorylated-ERK1/2 (p-ERK1/2), total ERK1/2, phosphorylated-GSK-3β (p-GSK-3β) and total GSK-3β expression in cerebellar granular cells (CGNs). CGNs were treated with 0.3 and 1 mM of Li and the cell lysates were prepared after 15, 30, 60 and 180 minutes. The protein expression was analyzed by western blotting. The p-ERK1/2 and ERK1/2 expression (A and B), and the p-GSK-3β and GSK-3β expression (A and C), were evaluated. The protein expression was normalized to β-actin as internal control (CTRL). The band intensity was measured and presented as the percent of un-treated cells (CTRL). All data were presented as Mean ± SD. Statistically significant values are presented as follows: *** p < 0.001, ** p < 0.01 and * p < 0.05, compared to CTRL; # p < 0.05, compared to respective time in two doses; † p < 0.001, compared the dose of 0.3 mM with the dose of 1mM at 60 min exposure
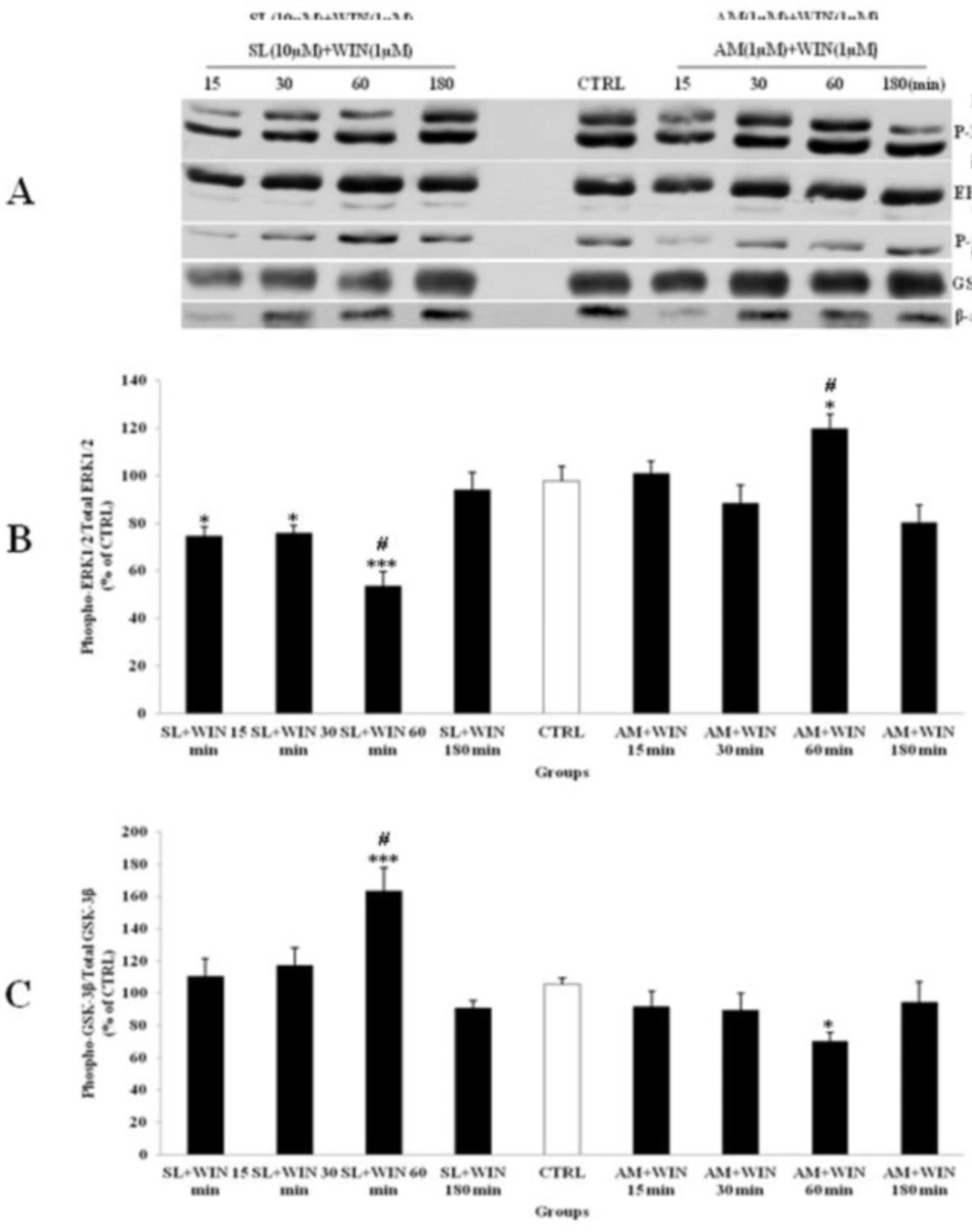
The effect of SL327 (SL) as a specific inhibitor of ERK1/2 pathway and AM251 (AM, 1 µM) as a cannabinoid antagonist on the phosphorylated-ERK1/2 (p-ERK1/2), total ERK1/2, phosphorylated-GSK-3β (p-GSK-3β) and total GSK-3β expression in cerebellar granular cells (CGNs). CGNs were pre-treated with SL (10 µM), 30 min before added WIN 55,212-2 (WIN, 1 µM) and AM was co-treated with WIN in CGNs 12-well plate and the cell lysates were prepared after 15, 30, 60 and 180 minutes. The protein expression was analyzed by western blotting. The p-ERK1/2 and ERK1/2 expression (A and B), and the p-GSK-3β and GSK-3β expression (A and C), were evaluated. The protein expression was normalized to β-actin as internal control (CTRL). The band intensity was measured and presented as the percent of un-treated cells (CTRL). All data were presented as Mean ± SD. Statistically significant values are presented as follows: *** p < 0.001, ** p < 0.01 and * p < 0.05, compared to CTRL; # p < 0.05, compared the 30 to 60 min exposure
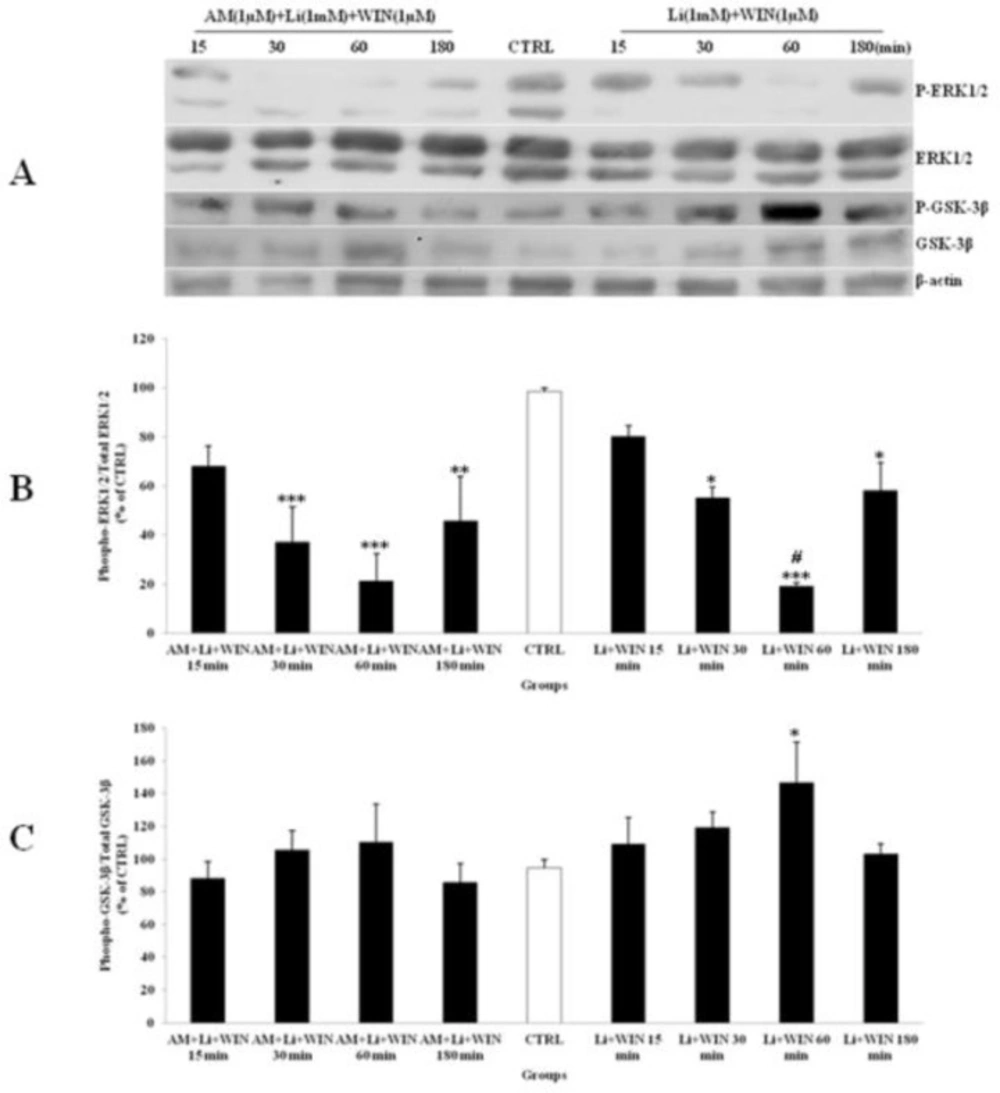
The effect of lithium (Li) pre-treated to WIN 55,212-2 (WIN) and AM251 (AM) on the phosphorylated-ERK1/2 (p-ERK1/2), total ERK1/2, phosphorylated-GSK-3β (p-GSK-3β) and total GSK-3β expression in cerebellar granular cells (CGNs). CGNs were pre-treated with Li (1mM), 30 min before added WIN (1 µM) and AM (1 µM), and the cell lysates were prepared after 15, 30, 60 and 180 minutes. The protein expression was analyzed by western blotting. The p-ERK1/2 and ERK1/2 expression (A and B), and the p-GSK-3β and GSK-3β expression (A and C), were evaluated. The protein expression was normalized to β-actin as internal control (CTRL). The band intensity was measured and presented as the percent of un-treated cells (CTRL). All data were presented as Mean ± SD. Statistically significant values are presented as follows: *** p < 0.001, ** p < 0.01 and * p < 0.05, compared to CTRL; # p < 0.05, compared the 30 to 60 min exposure
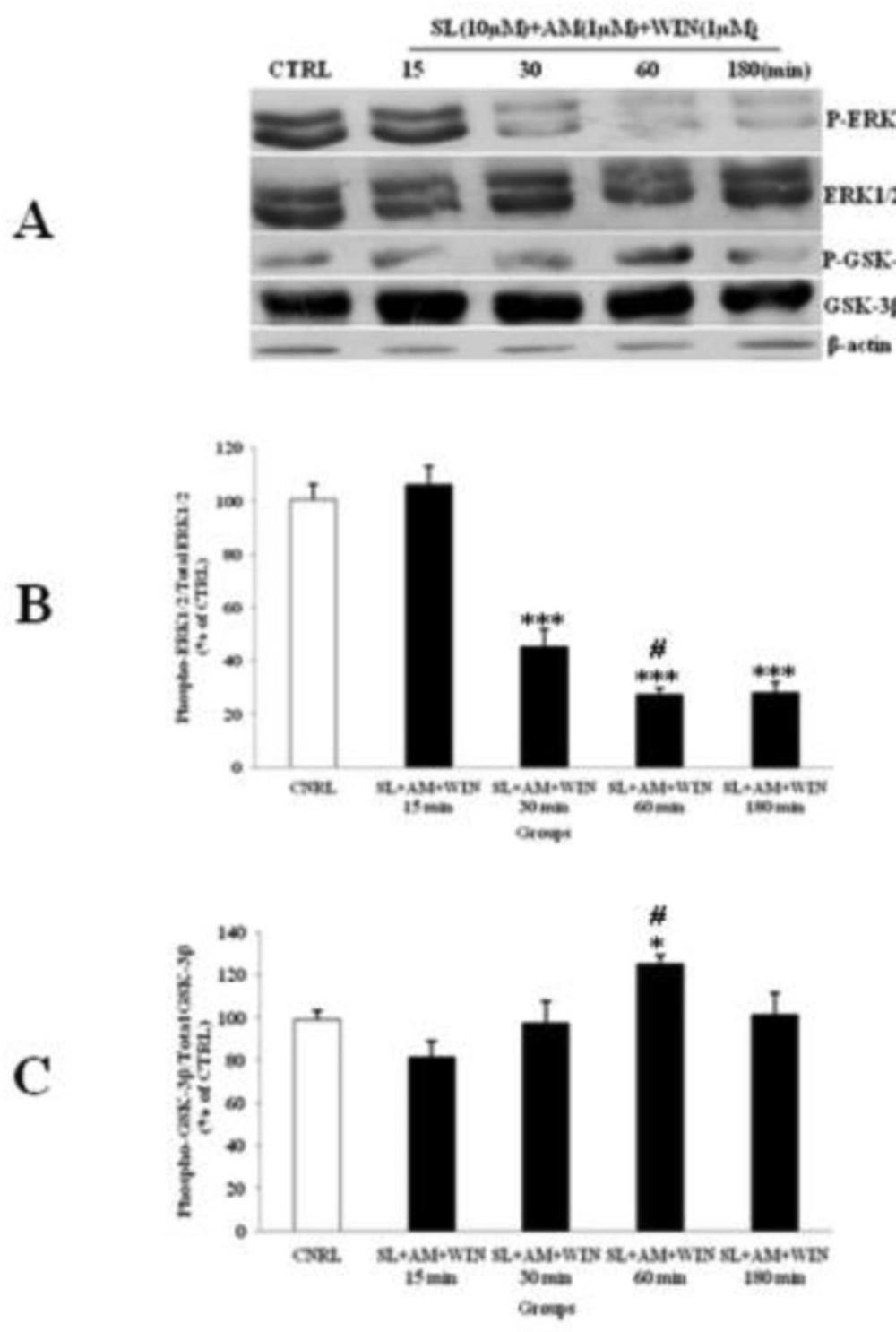
The effect of SL327 (SL) pre-treated to WIN 55, 212-2 (WIN) and AM251 (AM) on the phosphorylated-ERK1/2 (p-ERK1/2), total ERK1/2, phosphorylated-GSK-3β (p-GSK-3β) and total GSK-3β expression in cerebellar granular cells (CGNs). CGNs were pre-treated with SL (10 µM), 30 min before added WIN (1 µM) and AM (1 µM), and the cell lysates were prepared after 15, 30, 60 and 180 minutes. The protein expression was analyzed by western blotting. The p-ERK1/2 and ERK1/2 expression (A and B), and the p-GSK-3β and GSK-3β expression (A and C), were evaluated. The protein expression was normalized to β-actin as internal control (CTRL). The band intensity was measured and presented as the percent of un-treated cells (CTRL). All data were presented as Mean ± SD. Statistically significant values are presented as follows: *** p < 0.001, ** p < 0.01 and * p < 0.05, compared to CTRL; # p < 0.05, compared the 30 to 60 min exposure
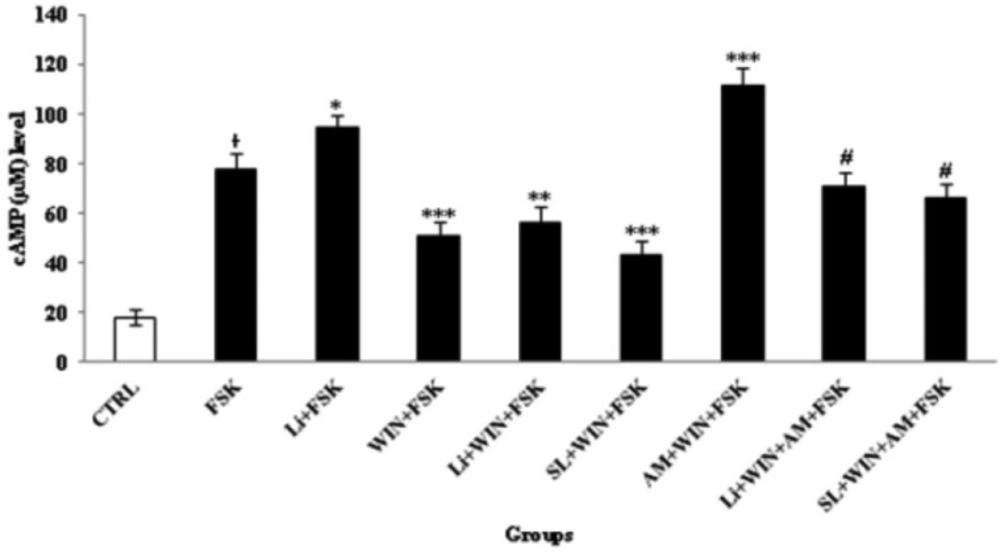
Cyclic AMP (cAMP) changes in study-treated groups in cerebellar granular neurons (CGNs). CGNs were cultured at 3.2 × 105/cm2 density in 12-well plate and treated with described method for 60 min. Before, the cAMP measurement, forskolin (FSK, 1 µM) was added for 10 min following the addition of WIN 55,212-2 (WIN, 1 µM) or Lithium (Li, 1 mM) into the CGNs medium, for induction of cAMP level. The control (CTRL) group was CGNs without treatment. All data were presented as Mean ± SD. Statistically significant values are presented as follows: * p < 0.05, compared to CTRL, *** p < 0.001, ** p < 0.01 and * p < 0.05, compared to FSK group; # p<0.05, compared to AM (AM251, 1 µM) + WIN 55, 212-2 (WIN, 1 µM) + FSK (1 µM) group

