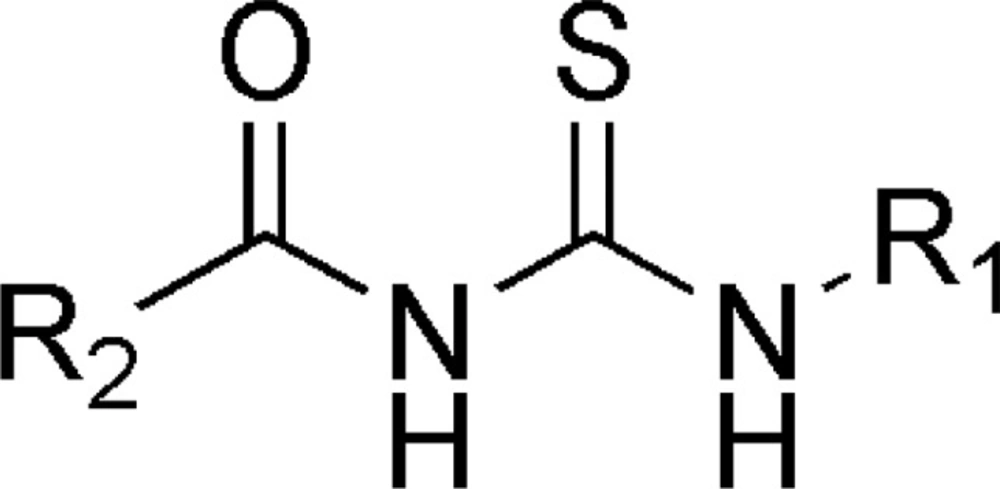
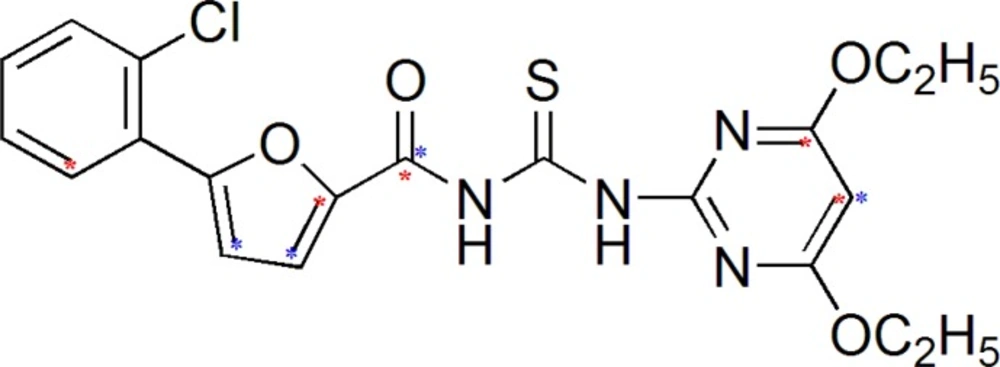
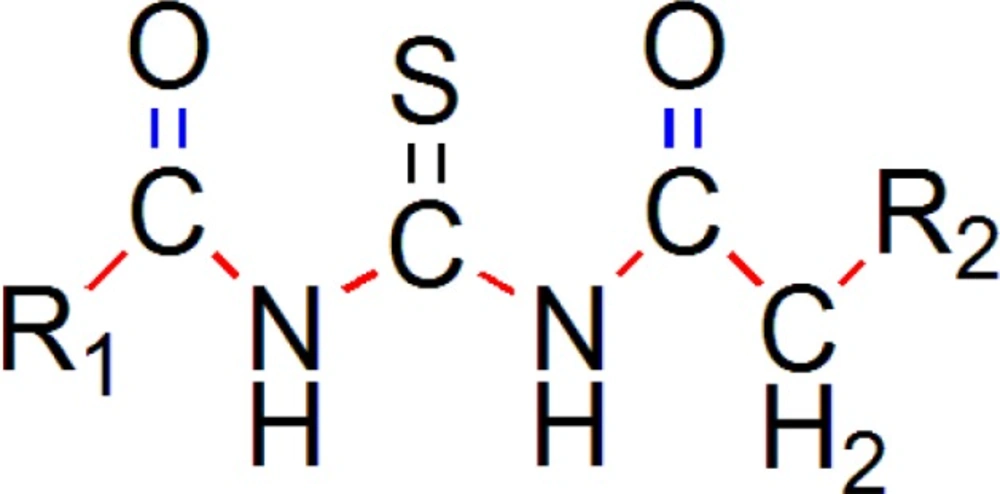
Result of 3D-QSAR
Series of Acyl thiourea derivatives was carefully divided into training set and test set. 3D-QSAR Studies were carried out by k Nearest Neighbor Molecular Field Analysis (kNN MFA) on the selective training set and test set as depicted in
Table 2.
1) Training set molecules
|
|---|
| Sr.No | R1 | R2 | IC50 | pIC50 |
|---|
| 1 | 4-Ethoxy-6-Methyl pyrimidine | 5-(2-chlorophenyl)-2-furyl | 1.65 | -0.21748 |
| 2 | 4,6-diethoxy pyrimidine | 5-(2-chlorophenyl)-2-furyl | 0.08 | 1.09691 |
| 3 | 4-Hydroxy-6-Methyl pyrimidine | 5-(2-chlorophenyl)-2-furyl | 0.32 | 0.49485 |
| 4 | 4,6-dimethoxy pyrimidine | 5-(2-chlorophenyl)-2-furyl | 1.77 | -0.24797 |
| 5 | 4,6-dichloro pyrimidine | 5-(2-chlorophenyl)-2-furyl | 14.5 | -1.16137 |
| 6 | 4,6-dichloro pyrimidine | 5-(4-nitrophenyl)-2-furyl | 1.66 | -0.22011 |
| 7 | t-butylamino carbonyl | 5-(4-nitrophenyl)-2-furyl | 1.30 | -0.11394 |
| 8 | t-butylamino carbonyl | Phenyl | 1.79 | -0.25285 |
| 9 | t-butylamino carbonyl | Methyl | 1.83 | -0.26245 |
| 10 | t-butylamino carbonyl | (2,4-Dichloro-Phenyl)-OCH2 | 1.67 | -0.22272 |
| 11 | t-butylamino carbonyl | 2,6-Difluoro-Phenyl | 1.43 | -0.15534 |
| 12 | t-butylamino carbonyl | 2-Methyl-1-(4-Chloro-Phenyl)-Propane | 1.35 | -0.13033 |
| 13 | t-butylamino carbonyl | 3-(2, 2-dichloro ethenyl)-2,2-dimethyl cyclopropyl. | 0.26 | 0.58503 |
| 14 | 4-Methoxy-6-chloro pyrimidine | 5-(2-chlorophenyl)-2-furyl | 1.29 | -0.11059 |
| 15 | 4-Methyl-6-Hydroxy pyrimidine | 6-Chloro-3-Pyridine | 8.58 | -0.93349 |
| 16 | 4,6-dimethoxy pyrimidine | 5,6-Dichloro-3- Pyridine | 18.5 | -1.26717 |
| 17 | 4,6-dimethyl pyrimidine | Phenyl | 2.1 | -0.32222 |
| 18 | 4,6-diethoxy pyrimidine | 2-Methyl-1-(4-Chloro-Phenyl)-Propane | 0.31 | 0.50864 |
| 19 | 4,6-dimethoxy pyrimidine | 3-(2-chloro-3,3,3-trifluropropenyl)-2,2-dimethyl cyclopropyl | 0.97 | 0.01323 |
| 20 | 4,6-dimethyl pyrimidine | 3-(2-chloro-3,3,3-trifluropropenyl)-2,2-dimethyl cyclopropyl | 0.58 | 0.23657 |
| 21 | 4,6-dimethyl pyrimidine | 2-Fluoro-4-Chloro-Phenyl | 1.36 | -0.133539 |
| 22 | 4-Methoxy-6-chloro pyrimidine | 2-Fluoro-4-Chloro-Phenyl | 5.1 | -0.70757 |
| 2) Test set molecules. |
| 1 | 4-Hydroxy-6-Methyl pyrimidine | 5-(4-nitrophenyl)-2-furyl | 0.36 | 0.4437 |
| 2 | t-butylamino carbonyl | 5-(2-Chloro-Phenyl)-2-Furyl | 1.42 | -0.152288 |
| 3 | t-butylamino carbonyl | 3-(2-chloro-3,3,3-trifluropropenyl)-2,2-dimethyl cyclopropyl | 0.51 | 0.29243 |
| 4 | 4-Methoxy-6-methyl pyrimidine | 5-(4-nitrophenyl)-2-furyl | 1.22 | -0.08636 |
| 5 | 4,6-dimethyl pyrimidine | 2-Chloro-3- Pyridine | 7.19 | -0.856729 |
| 6 | 4,6-dimethoxy pyrimidine | (2,4-Dichlorophenyl)-OCH2 | 1.89 | -0.276462 |
| q2 | pred_-r2 | q2- se | K Nearest Neighbour | Pred- r2se |
|---|
| 0.6840 | 0.7697 | 0.2811 | 2 | 0.2972 |
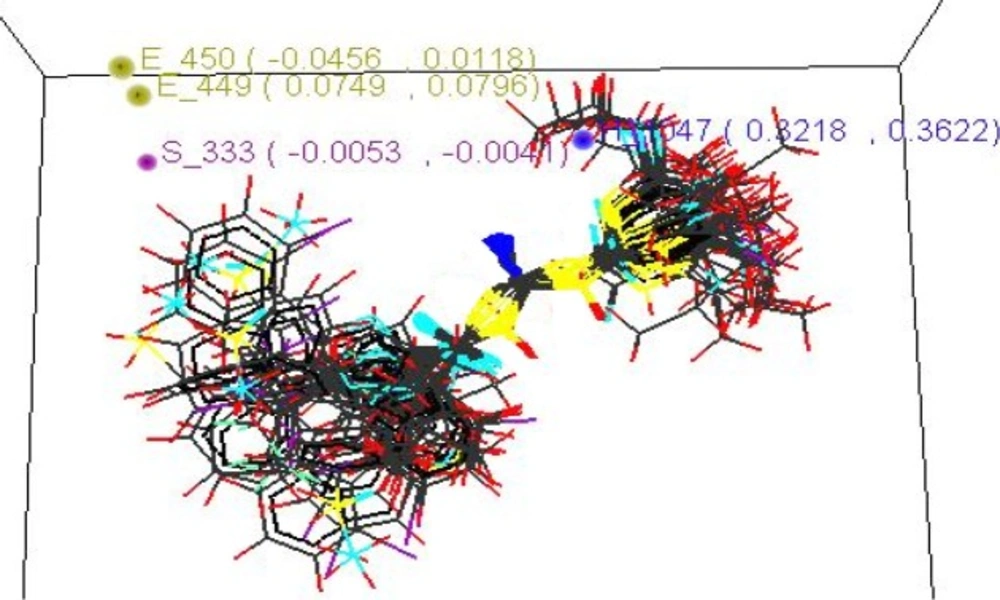
| ContributingStericParameters | S-333(-0.0053,0.0041) |
| ContributingElectronicParameters | E-449(0.0749,0.0796)E-450(-0.0456,0.0118) |
| ContributingHydrophobicParameters | H-1047(0.3218,0.3622) |
| N | 24 |
kNN MFA is a novel method which utilizes k-Nearest Neighbor (kNN) principle to correlate molecular field descriptors with biological activity. The kNN methodology relies upon a simple distance learning approach. In this method an unknown member is classified according to the majority of its k-Nearest Neighbors in the training set. The nearness is measured by an appropriate distance metrics (e.g., a molecular similarity measure calculated using field interactions of molecular structures). The standard kNN Method is implemented simply as follows:
The distances between an unknown object (u) and all other objects in the training set were calculated.
The k objects were selected from the training set most similar to object u, according to the calculated distances.
3. The object u was classified with the group to which the majority of the k objects belong. An optimal k-value is selected by optimization through the classification of a test set of samples or by Leave-One Out (LOO) cross validation (
19). The variables and optimal k-values were chosen using different variable selection methods. Here we have used simulated annealing as variable selection method.
Statistical result of 3D-QSAR kNN MFA method is tabulated below (
Table 3).
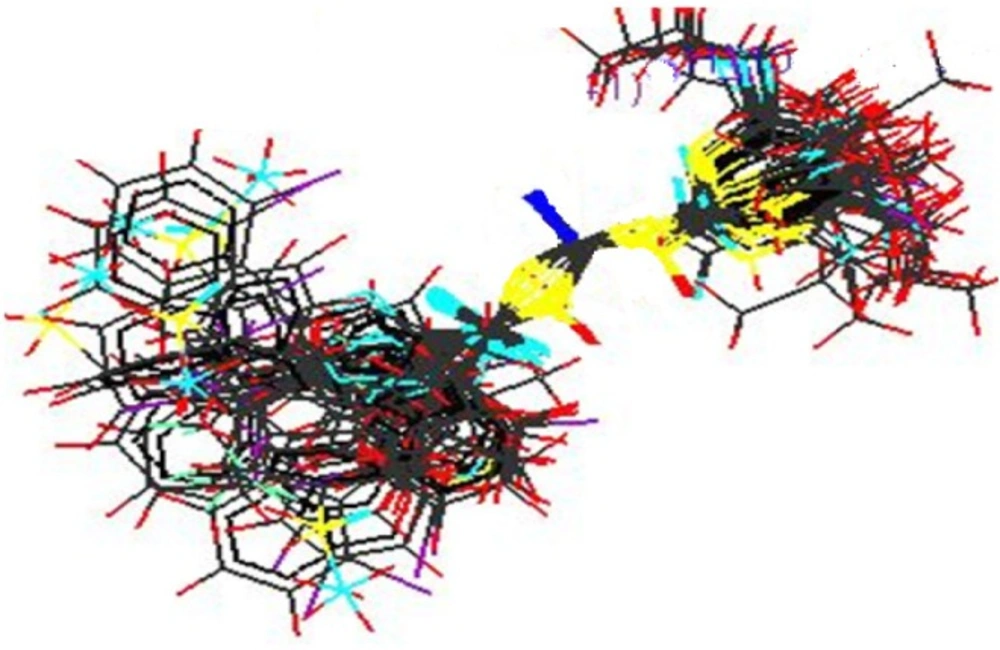
Stereo view of the molecular rectangular field grid generated around the superposed molecular units of Acyl Thiourea series using SA kNN-MFA Model
| MolNo. | R1 | R2 | Predicted activity | Lipinski’s scoreandScreen |
|---|
| R-1 | 4-nitrophenyl | 4-methoxyphenyl | 0.8564 | ADRXWS (6) |
| R-2 | 4-nitrophenyl | 4-nitrophenyl | -0.3876 | ADRXWS (6) |
| R-3 | Benzyl | 4-hydroxyphenyl | 0.9765 | ADRXWS (6) |
| R-4 | 3-aminophenyl | 4-nitrophenyl | 0.8450 | ADRXWS (6) |
Model showed best internal as well as external predictivity as q
2 = 0.6840, pred_r
2 = 0.7697 and also error occurred during internal and external validation obtained was very low as q
2_se = 0.2811, pred_r
2_se = 0.2972.Stereo view of superposed molecular units of Acyl Thiourea series shows the generated data points around the pharmacophore in three dimensional manners, which is shown below (
Figure 5).
Stereo view of the molecular rectangular field grid generated around the superposed molecular units of Acyl Thiourea series using SA kNN-MFA Model
Thus, SA KNN-MFA model leads to identification of various local interacting molecular features responsible for activity variation and hence provides direction for design of new molecules in a convenient way.
Interpretation from 3D QSAR Studies and Correlation of 3D descriptors
Electronic Parameters
3D QSAR studies revealed the electronic requirements around the acyl thiourea pharmacophore. The points those were found in SA KNN-MFA model are E_449, E_450 implying that these points are indeed significant for structure activity relationship and require the electronic properties as mentioned in the ranges in parenthesis for maximum biological activity.
Range for electronic descriptor E_450 (-0.0456, 0.0118).Descriptor ranging from negative to positive values specifies that the pharmacophore should be substituted with electronegative and electropositive groups so that it should increase the neuraminidase inhibiting activity, such as –OH, –Cl, -NO2, -OR and most potent compound possesses two electronegative groups such as –Cl, -OC2H5 from the parent series.
Steric Parameters
The steric points that were found in SA KNN-MFA model is S_333 implying that these points are indeed significant for structure activity relationship and require the steric properties as mentioned in the ranges in parenthesis for maximum biological activity. Range for steric descriptor is S_333 (-0.0053, -0.0041). Descriptor showing negative values specifies that the pharmacophore should be substituted with sterically less bulky group so that it can increase the NA inhibiting activity, such as lower alkyl groups, unsubstituted or mono substituted aryl rings.
Study of Structural Necessity for Neuraminidase Inhibition
Wang and Wade have reported guidelines for structural modification of developing neuraminidase inhibitors and the design of novel inhibitors in order to optimize inhibitory activity (
Figure 6) (
23). Since the catalytic site of influenza virus Neuraminidase was totally conserved among all influenza viral strains, an ideal drug which effectively blocked one neuraminidase would be effective at blocking all other neuraminidases, even those on viruses which have not yet appeared in humans (
8).
Thus, along with QSAR study, we are herein considering the essential structural necessities which should be present in the acyl thiourea pharmacophore. It will be beneficial in such a way that we can design better compounds which would fit into neuraminidase binding site perfectly and would show best interactions than standard, marketed neuraminidase inhibitors.
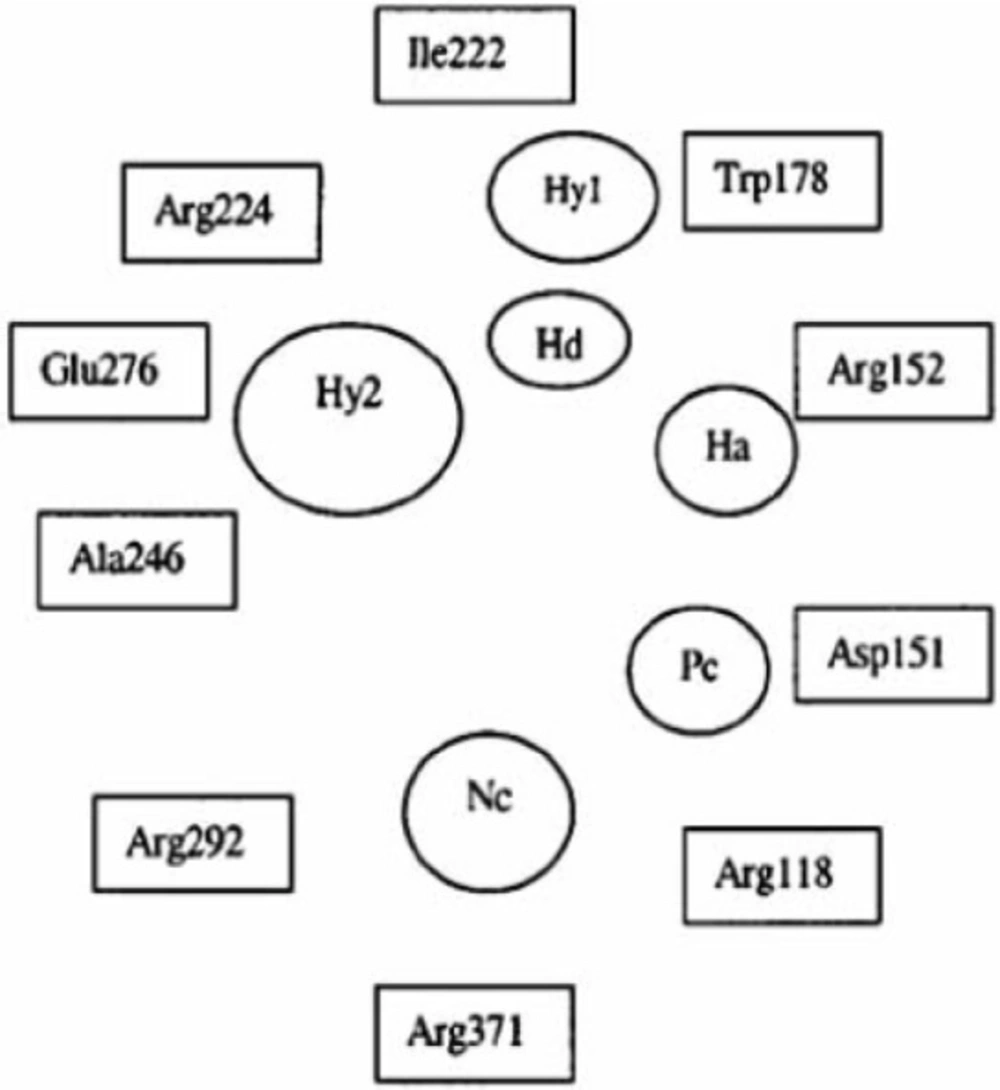
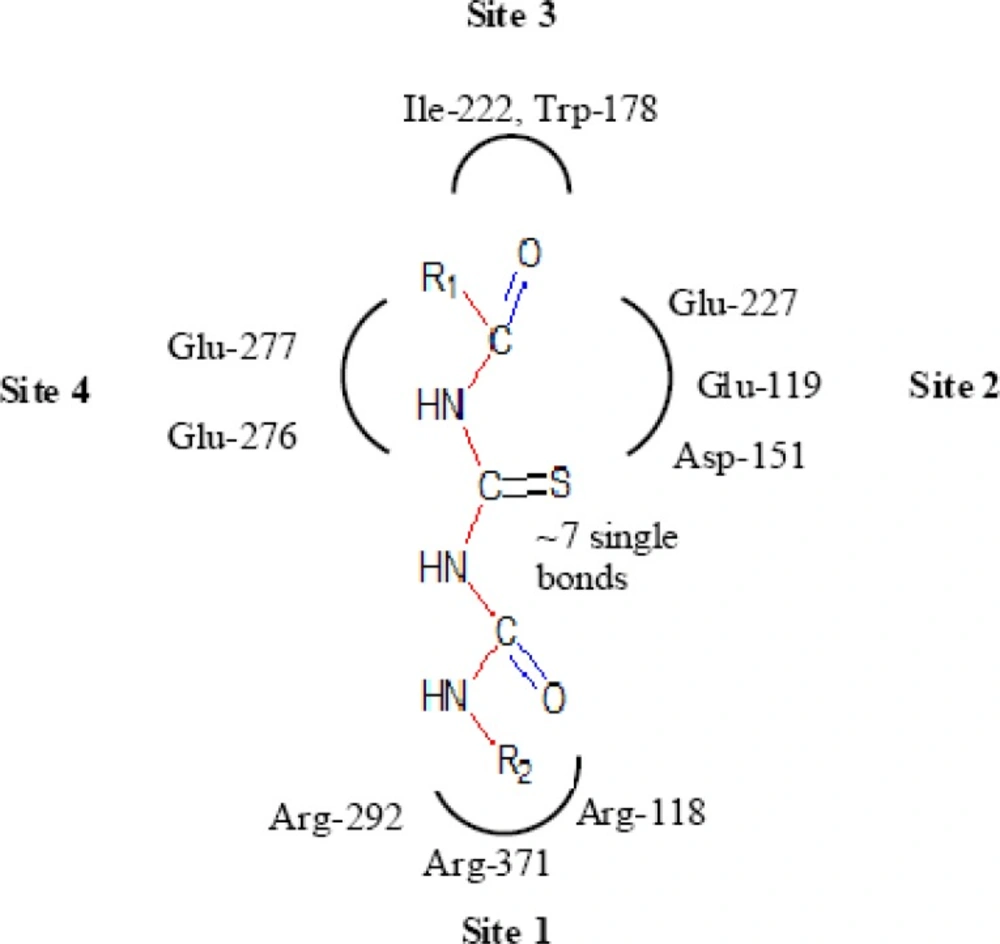
Important structural features for potent inhibitor and corresponding neuraminidase residues (23)
A negatively charged (Nc) group that makes strong charge-charge interactions with the triarginyl pocket as Arg 292, Arg 371 and Arg 118 is highly favorable for binding. Positively charged (Pc) group is favoured for binding by the electrostatic interactions with residue Asp151.While, Arg 152 requires hydrogen bond acceptor (Ha) group for binding. Trp 178 requires such a structural functionality which should be hydrogen bond donor (Hd) as well as small hydrophobic group for favourable binding. While, large hydrophobic group contributing to the van der Waals interaction should be preferred for three different amino acid residues such as Arg 224, Ala 246 and Glu 276.
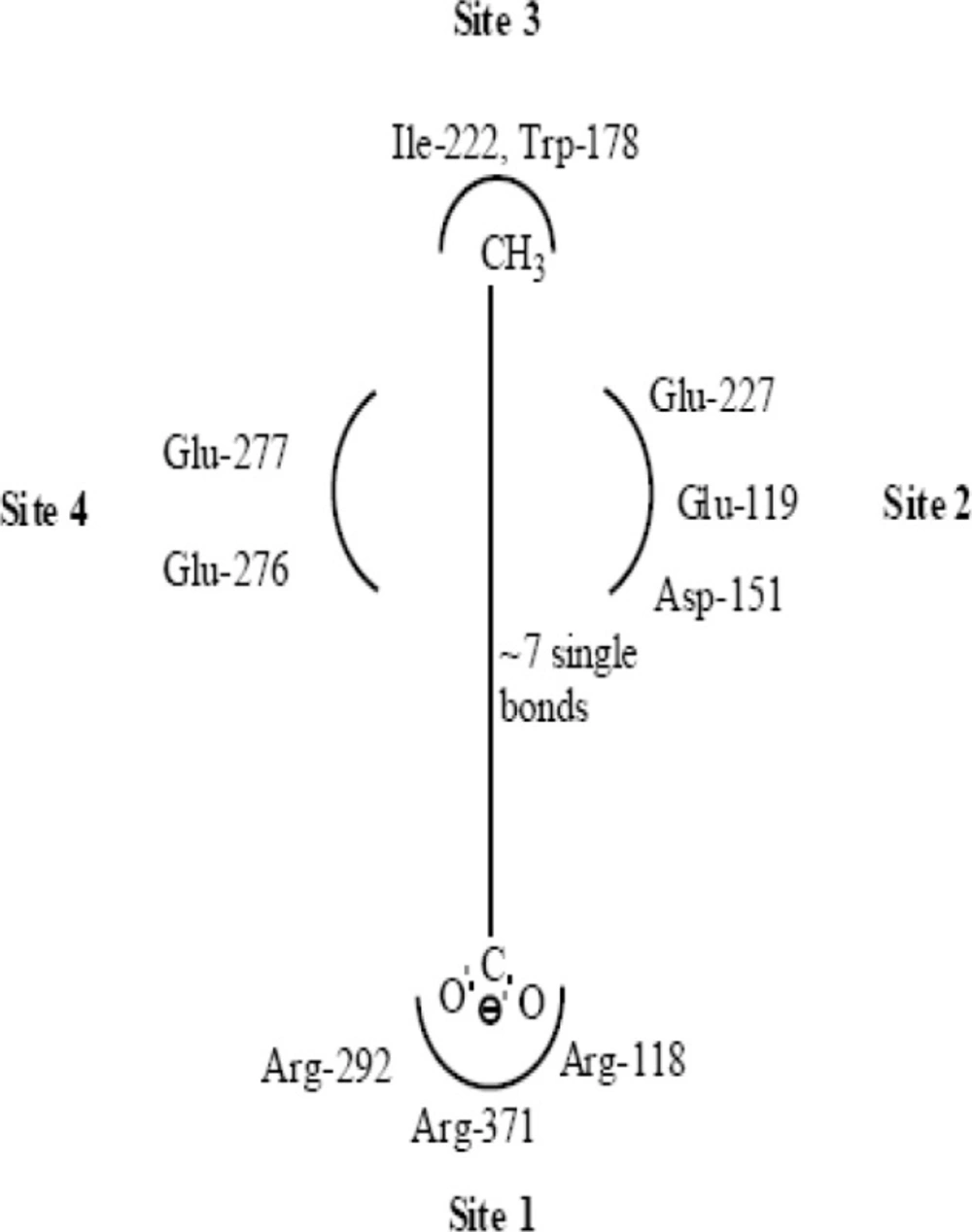
Along with structural necessities for neuraminidase inhibition, we are considering air plane model of neuraminidase enzyme, so as to design potential derivatives with excellent anti-influenza activity (
24). Wang and co-workers (
24) derived an airplane model of the neuraminidase active site as illustrated in (
Figure 7) to summarize the basic structural requirements of potent neuraminidase inhibitors. The active site of neuraminidase has four main well conserved binding sites. The centre of site 2 is about 6 A.U. from site 1 and about 4 A.U. from site 3, while site 4 is about 6 A.U. from site 1 and 5 A.U. from site 3. Sites 1 and 3 are separated by 9-10 A.U. or about 7 single bond lengths.
Diagram of neuraminidase sites S1-S4 and important nearby residues (25).
Gong and co-workers have proposed about the pocket study of neuraminidase active site (
8).
By taken into consideration, structural necessity and airplane model and pockets of neuraminidase enzyme, we will design such new chemical entities which are much potential than standard and selective inhibitors.
Design of New Chemical Entities
Designed compounds generated in this way were then screened by three type of screening methods; Lipinski’s rule and prediction of activity using multiple linear regression equation [pIC
50 = 0.250765 T_C_N_6 + 0.106694 T_C_O_2 + 0.637398 MMFF_2 - 0.623115 chiV3 + 6.1896] obtained by the two dimensional QSAR studies and to ensure drug like pharmacokinetic profile by prediction of ADMET properties for finding a new compounds having neuraminidase inhibitor and anti-influenza activity (
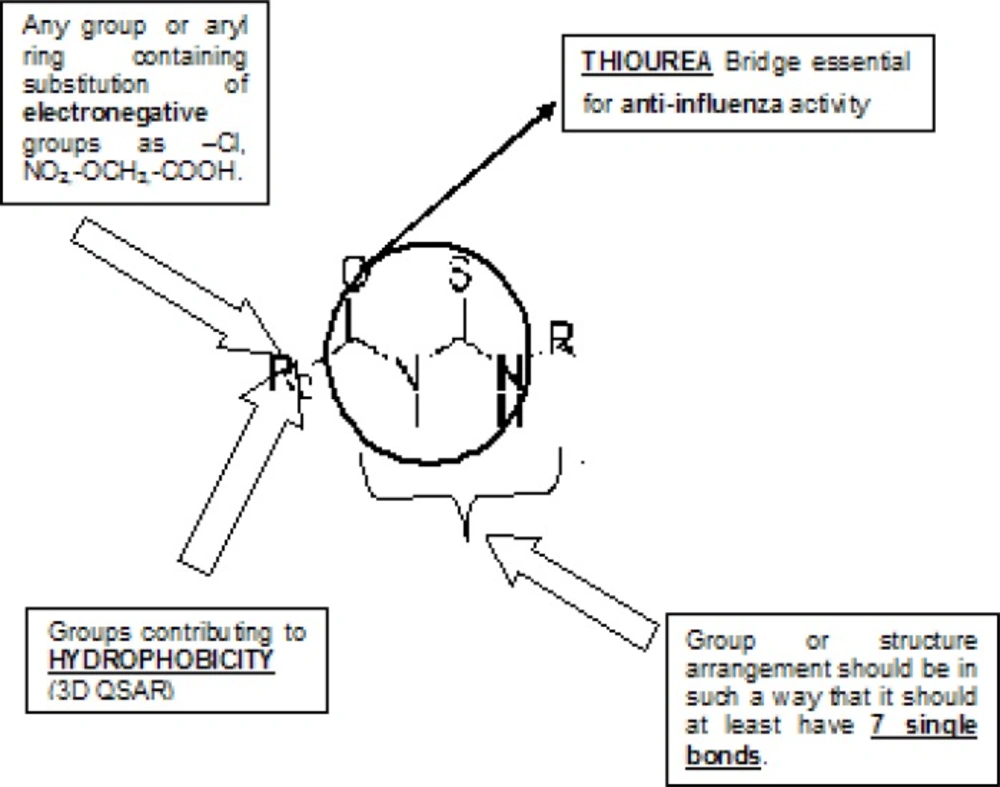
20). New chemical entities were generated using optimized pharmacophore shown in (
Figure 8).
Pharmacophore Requirement of Acyl thiourea Derivatives Generated for Selective Inhibition of neuraminidase for anti-influenza activity
More than one hundreds of molecules were generated using CombiLib tool which follows the Lipinski’s rule, but we have selected only 10 most active molecules on the basis of predicted activity. The most potent compounds have positive values of pIC50, similarly the predicted activity values for new chemical entities were found to lye towards positive side.Compounds qualifying all required parameters set for Lipinski’s Screen/filter are indicated by ADRXWS strings. The columns containing the Lipinski’s screen score and other column containing the strings of alphabets, ADRXWS indicate that all 6 conditions are satisfied by that corresponding compound. Lesser the screen score, lesser is the pharmacokinetic compatibility (drug likeliness) for that designed compound.
Generation of Templates
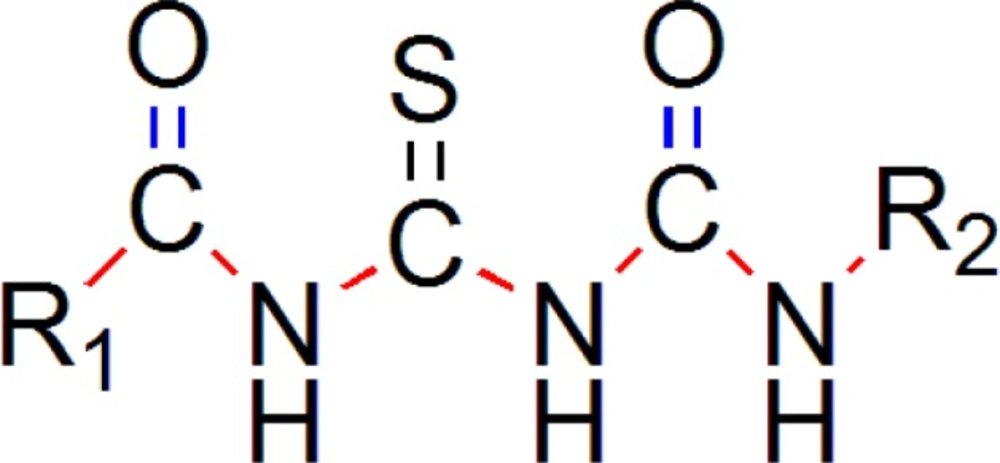
We have generated two templates considering above stated pharmacophore requirements. Designed templates consist of dicarbonyl groups which are different than parent series, while parent series has single acyl group in it, we have placed another acyl group instead of any aryl ring containing oxygen atom, as in parent series has furan ring in it (
19).
One template (
Figure 9) consists of 3 –NH groups in the pharmacophore, while another template (
Figure 10) consists of 2 –NH groups, one –NH group is isosterically replaced by –CH
2 group.
Both of the templates have seven single bonds between R
1 and R
2 substituents as it is basic structural requirement for neuraminidase inhibition, where in case of R
1 and R
2, we have used aryl ring with electronegative and electropositive groups such as–OH,-COOH,-NO
2 and –OCH
3, –NH
2 and aryl ring also contributes to hydrophobicity, as correlated in 3D-QSAR studies. Those seven single bonds are represented in red colour in designed template (
Figure 11). Even placement of extra carbonyl functionality and aryl ring with electronegative carboxyl group in the acyl thiourea pharmacophore becomes helpful for binding it with triarginyl residues as discussed above (
Figure 6,
7).
Designed template with active binding site
Generation of Combinatorial Library
We have generated more than hundrded molecules using CombiLib tool (
20) which follows the Lipinski’s rule, but we have selected only 10 most active molecules on the basis of their predicted activity using Multiple Linear regression equation.
We have selected only four molecules from the B template (
Table 4) considering promising predicted activity.While from A template we have selected seven molecules (
Table 5) on the basis of predicted activity.
| MoleculeNo. | R1 | R2 | Predicted activity | Lipinski’s score and Screen |
|---|
| R-5 | 3-nitro phenyl | 3-carboxyphenyl | -0.03206 | ADRXWS (6) |
| R-6 | 2-hydroxy phenyl | 3-carboxyphenyl | -0.06934 | ADRXWS (6) |
| R-7 | 2-hydroxy phenyl | 3-hydoxy phenyl | -0.156 | ADRXWS (6) |
| R-8 | 4-nitrophenyl | 3-carboxy phenyl | -0.18876 | ADRXWS (6) |
| R-9 | Benzyl | 3-hydoxy phenyl | -0.38875-0.17141 | ADRXWS (6) |
| R-10 | 4-methoxyphenyl | 3-carboxyphenyl | -0.39318 | ADRXWS (6) |
Out of total 11 designed new chemical entities only 3 compounds viz. R-8,R-9 and R-10 show predicted activity less potent as compared with most potent compound from the parent series with pIC50 1.09691,because higher the value of pIC50 more potent is the entity.
| MoleculeNo. | G-Score | E-model | No ofH aBond | No. ofGoodVdw b | No. ofBadVdw b | No. ofUglyVdw b |
|---|
| R-7 | -5.98 | -51.6 | 3 | 220 | 12 | 3 |
| R-8 | -5.58 | -50.5 | 4 | 233 | 10 | 3 |
| Peramivir | -5.53 | -45.5 | 7 | 278 | 14 | 2 |
| R-4 | -5.14 | -47.3 | 2 | 241 | 7 | 1 |
| R-10 | -5.07 | -43.2 | 8 | 150 | 9 | 2 |
| R-9 | -4.83 | -47.9 | 2 | 210 | 9 | 2 |
| R-2 | -4.79 | -41.2 | 3 | 234 | 11 | 1 |
| R-5 | -4.58 | -47..3 | 3 | 134 | 6 | 1 |
| Oseltamivir | -4.57 | -49.3 | 4 | 266 | 8 | 1 |
| R-3 | -4.27 | -51.3 | 2 | 233 | 6 | 0 |
| R-6 | -3.91 | -58.0 | 6 | 167 | 7 | 0 |
| R-1 | -2.74 | -2.74 | 2 | 173 | 6 | 0 |
Molecular Docking studies
Molecular Docking is a key tool in structural molecular biology and computer-assisted drug design. Selected most active molecules were docked on crystallographic structure of neuraminidase enzyme available in the RCSB PDB Database (Code: 3b7e) co-crystallized with the ligand Zanamivir (
27). A molecular docking study helps to determine possible interaction of new chemical entities with the enzyme on PDB (3b7e).
The goal of ligand–protein docking is to predict the predominant binding mode(s) of a ligand with a protein of known three-dimensional structure. Virtual screening on the basis of molecular descriptors and physicochemical properties of active ligands has great usefulness in finding hits and leads through library enrichment for screening (
28), a strategy that is also well-used for reducing and enriching the library of ligands for molecular docking; there are recent reports that ligand shape-matching does as well as, if not better than, docking (
29).
Glide was found to produce least number of inaccurate poses and 85% of Glides binding models had an RMSD of 1.4 A
0 or less from native co-crystallized structures (
30). The Glide docking program approximated a complete systematic search of the conformational, orientation and positional space of the docked ligand molecules in to the receptor (protein) binding pocket.
Overview of docking of New Chemical Entities
All the designed compounds that show good predicted activity and follows Lipinski’s rule were docked into neuraminidase enzyme (pdb code: 3b7e) to study the binding mode of designed compounds. Further screening to sort out the best compound having good binding affinity which was compared with binding mode of Standard Neuraminidase Inhibitors,
e.g. Oseltamivir, Zanamivir and Peramivir results of which are depicted in (
Table 6 and
7).
The reliability of the docking results was first checked by comparing the best docking poses obtained for the co-crystallized inhibitor with its bound conformation.
As a result, a root mean square deviation (RMSD) of 0.7 Å was found suggesting that the docking procedure could be relied on to predict the binding mode of our compounds.
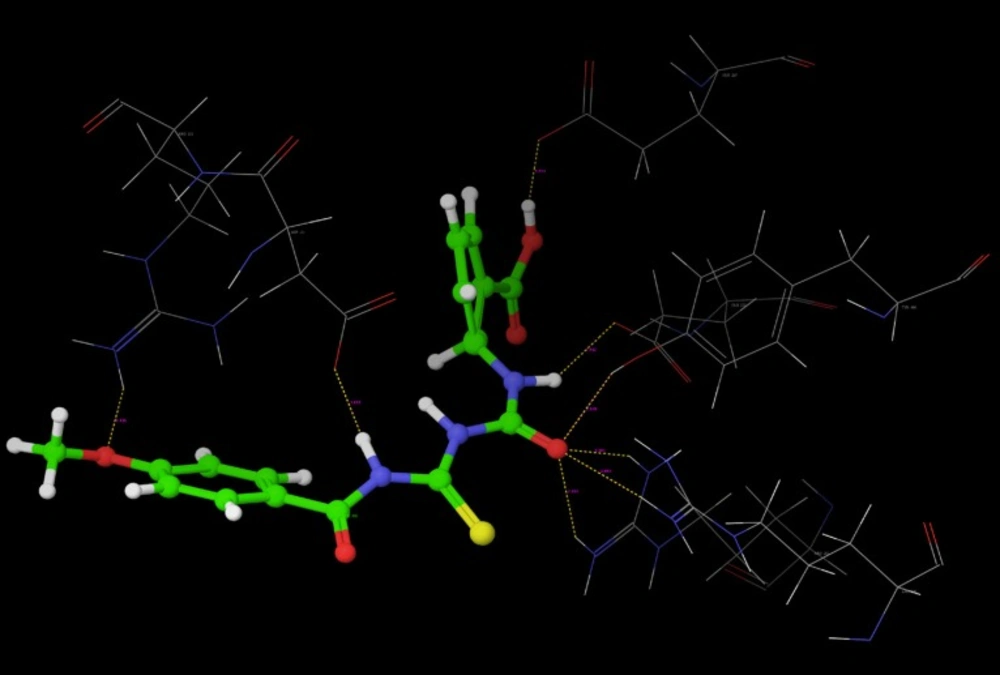
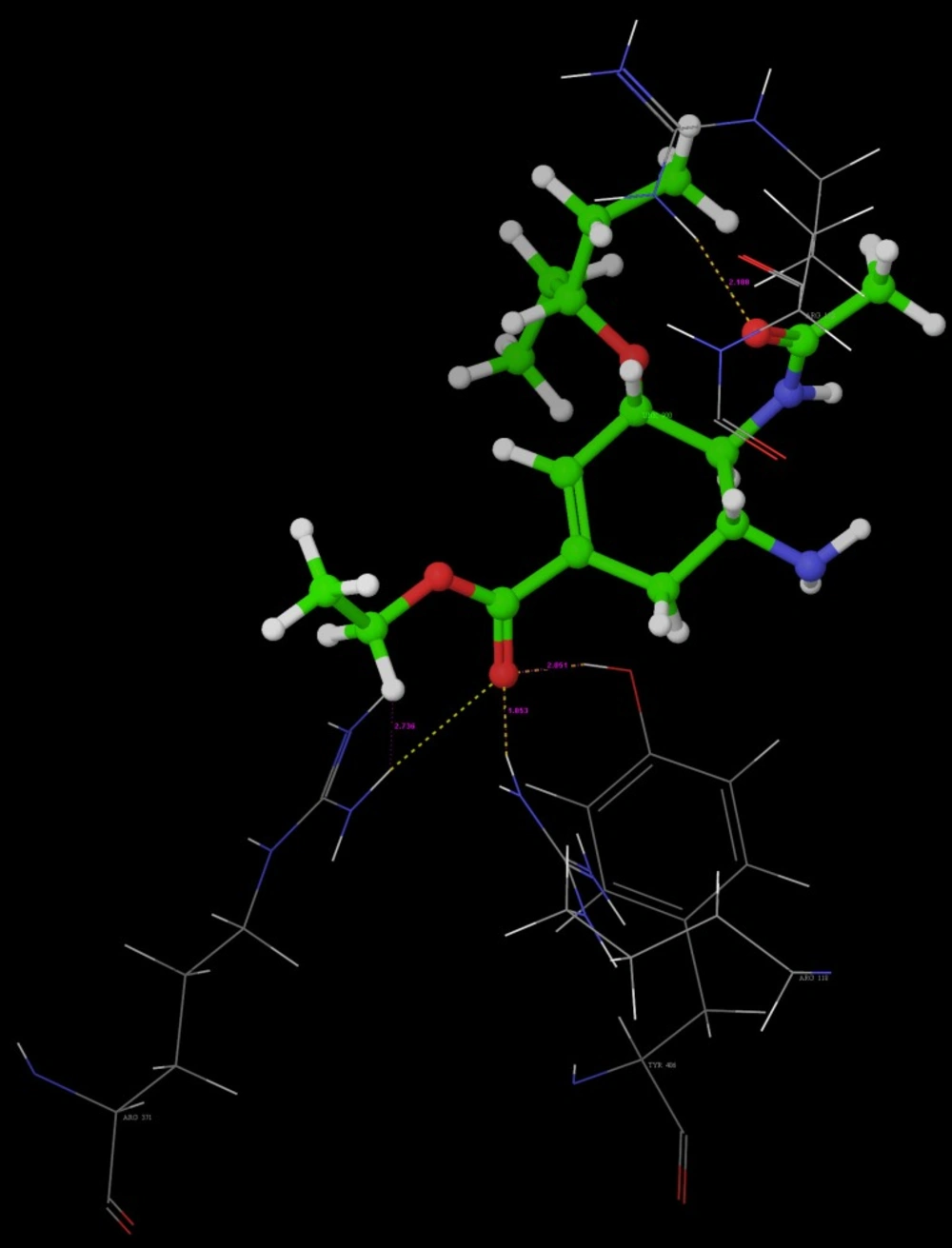
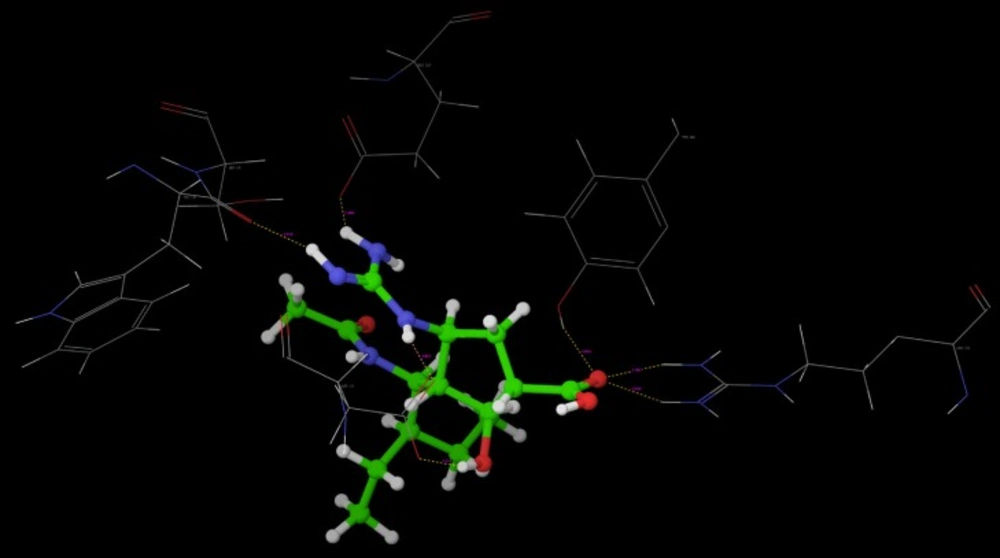
Poses of interactions of ligands along with standard are represented in (
Figure 12,
13,
14 and
15).
Hydrogen bond interaction of R-7
Hydrogen bond interaction of R-10
Hydrogen bond interaction of Oseltamivir
Hydrogen bond interaction of Peramivir
Key findings of overall Docking studies
The close inspection of result of molecular docking studies indicated that the designed compounds docked better than ligand Oseltamivir and Peramivir. Following are the reasons for the same.
1. G-Score
Molecules R-7 and R-8 show highest G-score than both the standards Peramivir and oseltamivir. While, molecules such as R-2, R-4, R-5, R-9 and R-10 show higher G-score than oseltamivir.
2. Hydrogen bond interactions
Molecule R-10 shows highest hydrogen bond interactions i.e. 8 than Peramivir and Oseltamivir, which show 7 and 4 interactions respectively. While, R-6 shows 6 hydrogen bond interactions which are more than oseltamivir.
3. Van der Waals interactions
R-1, R-3, R-4, R-5, R-6 show less bad van der Waals interactions than oseltamivir and Peramivir, where only Peramivir shows highest bad van der Waals interactions as 14 and oseltamivir shows 8 bad van der Waals interactions. Molecules R-1, R-3 and R-6 show zero ugly forces, where oseltamivir and Peramivir show 2 and 1, respectively.
4. In all the bad and ugly contacts penalize the G-score and overall conformation of ligand does matter a lot while binding to amino acid residues at active binding site. This also penalizes the energy of the model.
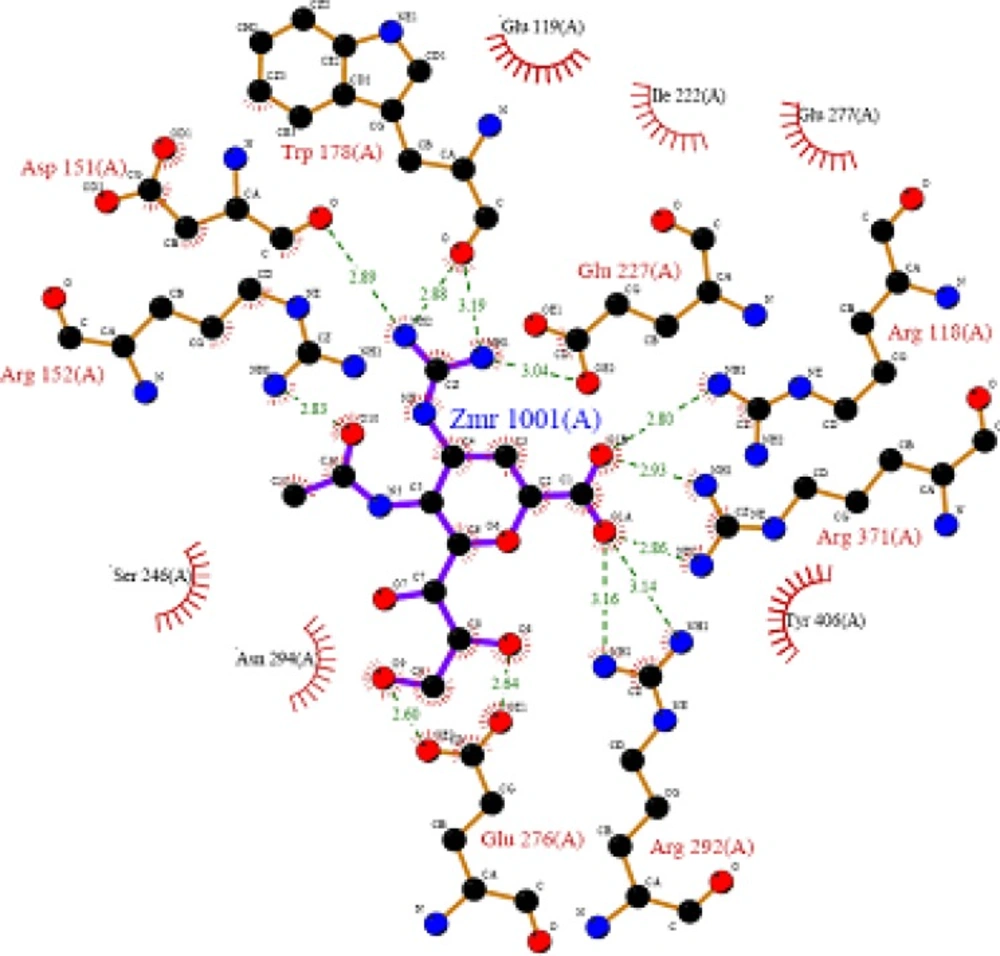
Key interactions are depicted in tabular form (
Table 7). It was observed from docking studies that all ligands lie in the same pocket of neuraminidase binding site of enzyme containing Arg 152, Tyr 406, Arg 371, Arg 118, Glu 227, Glu 277, Asp 151, Arg 292, Asn 294 and Ser 246 amino acids. Thus designed compounds showed a good binding affinity for interaction with neuraminidase active binding site.R-7, R-8, R-9 and R-10 show Hydrogen bond interaction with different arginine residues, in this case one carbonyl group, a negatively charged group from dicarbonyl thiourea interacts with positively charged groups or ions such as –NH
2 or H
+ of OH arginine residues. Here, one of structural necessity for selective neuraminidase inhibition is satisfied.
R-7, R-8, R-9 and R-10 show electrostatic interaction with different glutamic acid residues. In their case, either a positively charged group such as –NH2 or positively charged ion H+ of –OH or –COOH interacts with carbonyl or carboxyl group of glutamic acid residues, satisfying structural necessity for selective neuraminidase inhibitor.
Oseltamivir shows Hydrogen bonding with amino acids Arg 152, Tyr 406, Arg 371, Arg 118 which are one of essential interactions for neuraminidase inhibition, which is described previously in study of structural necessity for neuraminidase inhibition. Distance of hydrogen bonds in NCEs and standard compounds also explain better interaction (
Table 7) with neuraminidase binding site in 3b7e pdb.
The hydrogen bond distance in case of compound R-7 with Glu 227 (1.793 A.U.) which is less as compared to the hydrogen bonding interaction in case of Peramivir which is 2.477 A.U. Hence, less hydrogen bonding distance than standards is required for better binding which is reported in NCEs during docking study.
G-score is calculated taking all above aspects in to consideration, while designing new chemical entities to inhibit Neuraminidase more effectively and in turn to optimize the pharmacophore required for selective inhibition of Neuraminidase.
| Molecule No. | H-bond interactionWith amino acid | H-bond interactionin A.U. | Functional Group of structure involved in bonding | Amino acid Residue involved in bonding |
|---|
| R-7 | Glu 119Arg 118Glu 227 | 1.7931.8071.779 | -NH-C=OH of OH | -COOH-NH2-COOH |
| R-8 | Glu 119Arg 118Glu 227Tyr 406 | 1.7471.9131.7381.929 | -NH-C=OH of COOH-C=O | -COOHH of OH-COOH-NH2 |
| Peramivir | Trp 178Glu 227Tyr 406Asp 151(2)Arg 371(2) | 1.9602.4771.985a) 1.803b) 2.315a) 2.207b) 1.966 | -NH of guanidine-NH of guanidine-C=O of COCH3a) -NH of guanidineb)H of OHa) -C=O of COCH3b) -C=O of COCH3 | -C=OH of OHH of OHa)-C=Ob) -C=O-NH2-NH2 |
| R-4 | Asp 151Trp 178 | 2.0681.976 | -NH-NH | -C=O-C=O |
| R-10 | Asp 151Arg 292Arg 371(2)Glu 277Tyr 406Glu 227Arg 152 | 1.8512.992a)2.353b)2.1012.4822.1791.7312.135 | -NH-C=Oa) -C=Ob) -C=O-NH-C=OH of OHO of OCH3 | -C=O-NHa)-NHb)-NH-C=OH of OH-C=O-NH |
| R-9 | Glu 119Arg 118 | 1.9381.894 | -NH-C=O | -C=O-NH |
| R-2 | Asn 294Ser 246Asn 221 | 2.32.1022.138 | O of NO2O of NO2O of NO2 | -NH-NH-NH |
| R-5 | Asp 151Tyr 406 | 2.2132.412 | -NH-C=O | -C=OH of OH |
| Oseltamivir | Arg 152Tyr 406Arg 371Arg 118 | 2.182.0512.4701.853 | -C=O of COCH3-C=O of COCH3-C=O of COOC2H5-C=O of COOC2H5 | -NH-OH-NH-NH |
ADMET Prediction
Prediction of ADMET properties was used as last screen to sort out those compounds that already follow Lipinski’s rule and show good predicted activity and binding conformation at Neuraminidase receptor.
The parameters illustrated in (
Table 8) Qikprop analysis show significant results. CNS parameter is related with absorption of entity through Blood brain barrier, standard limit for CNS is -2 to +2, where -2 shows inactive CNS penetration and +2 shows active CNS penetration. All the designed entities show satisfactory results, with negative values, indicating poor CNS penetration. % Oral Absorption parameter is related with extent of oral absorption of drug, indicating suitable route of administration, if it is going to be formulated. If entity shows more than 80% oral absorption, it is considered to be highly absorbed. While if any entity shows less than 25% oral absorption, it is considered to be poorly absorbed. Metabolites suggest the number of metabolites which will possibly generate after undergoing metabolic changes, number of metabolites should range from 1-8.
| Molecule No. | Molecularweight | CNS | % OralAbsorption | No.of possible metabolites |
|---|
| R-7 | 331.345 | -2 | 78.416 | 3 |
| R-8 | 388.354 | -2 | 48.485 | 2 |
| Peramivir | 328.411 | -2 | 29.001 | 3 |
| R-4 | 358.371 | -2 | 73.409 | 4 |
| R-10 | 373.383 | -2 | 73.351 | 2 |
| R-9 | 329.373 | -2 | 84.192 | 3 |
| R-2 | 388.354 | -2 | 68.372 | 3 |
| R-5 | 358.371 | -2 | 43.878 | 3 |
| Oseltamivir | 312.408 | -1 | 68.391 | 3 |