Cell culture and treatment
MCF-7(noninvasive human breast cancer cell line, ER-positive) and MDA-MB-231(invasive and metastatic human breast cancer cell line, ER-negative) were grown in RPMI 1640 media supplemented with 10% (v/v) fetal calf serum(FCS) and penicillin/streptomycin antibiotics. Cultures were maintained at 37 °C in a humidified incubator containing 5% CO2.
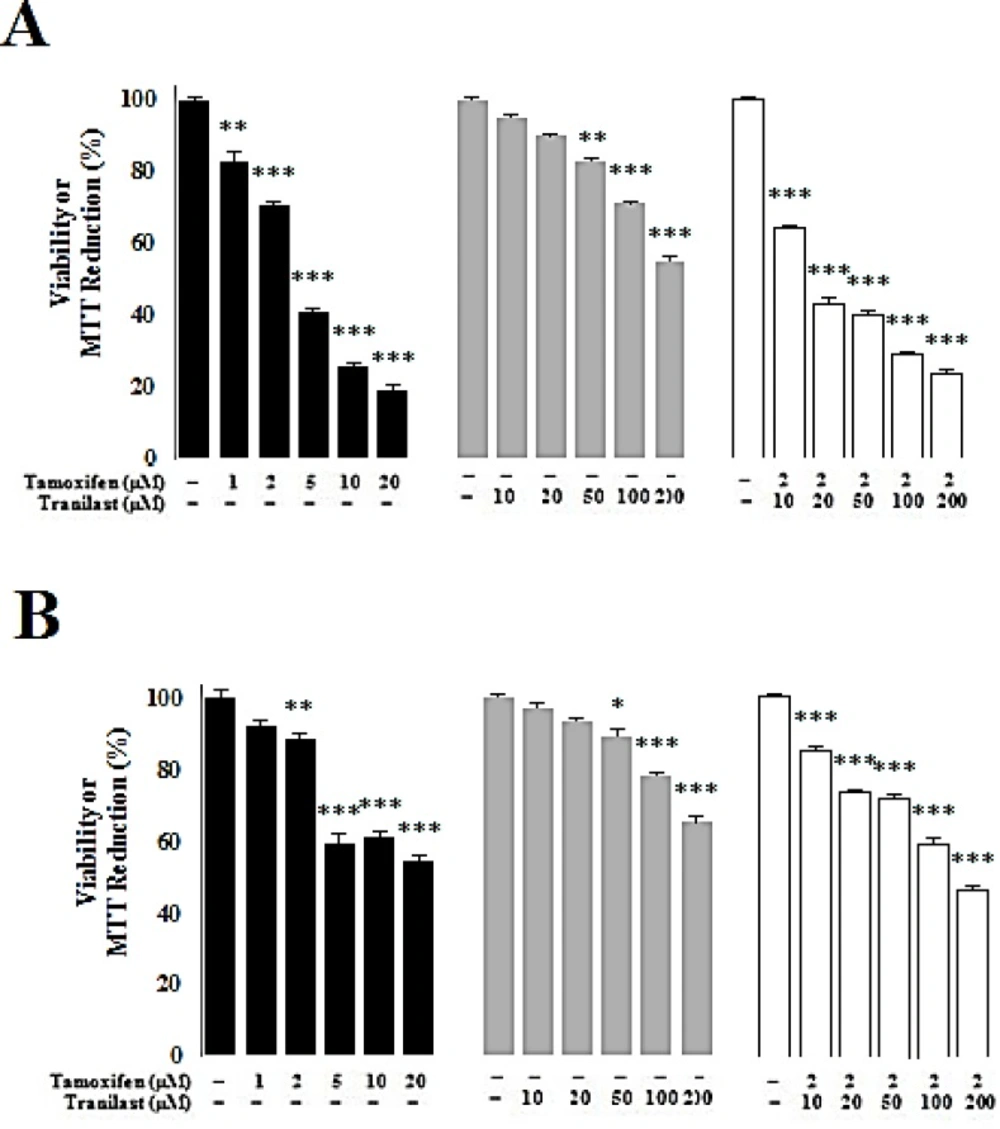
Tamoxifen (TAM) and tranilast (TRAN) were procured from Enzo Life Sciences and dissolved in dimethyl sulfoxide. Single treatments of TAM were 1, 2, 5, 10, 20 µM and for, TRAN were 10, 20, 50, 100, 200 µM as a single treatment. In the combination treatment, it was used 2 µM of TAM with 10, 20, 50, 100, 200 µM of TRAN.
Cell viability assay
MCF-7 or MDA-MB-231 cells were seeded at 104 cells/well in 96-well culture plates. All drug concentrations were tested in triplicate wells and the assays were performed in three separate experiments. After incubation for 48 h at 37 °C and 5% CO2, 20 μL of MTT solution (5 mg/mL in PBS) was added to each well and incubated for 4h at 37 °C. The medium with MTT were removed, and 100 μL of dimethyl sulfoxide was added to dissolve formazan crystal at room temperature for 30 min. The optical density (OD) of each well was measured by plate reader at 570 nm.
In-vitro wound assay
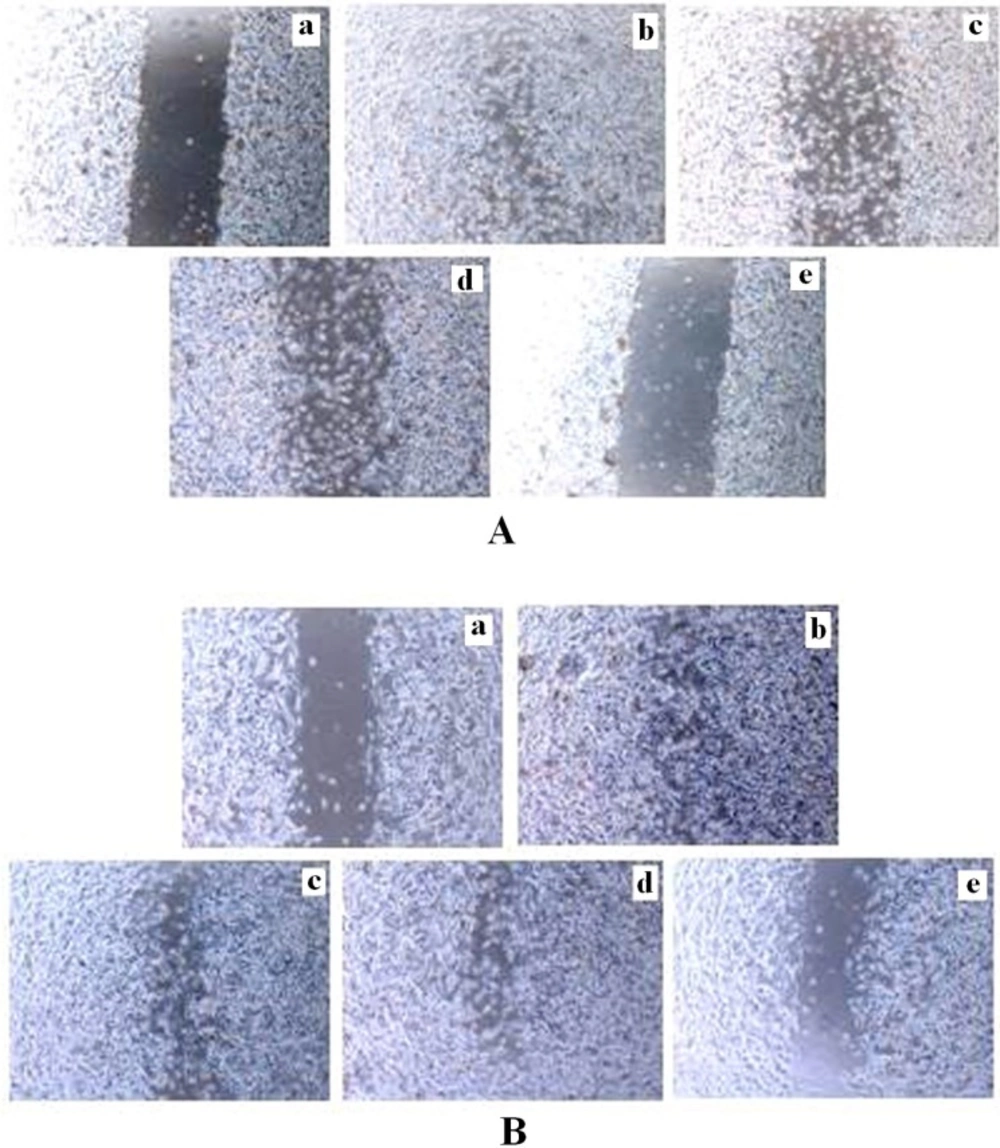
In-vitro wound assay for studying of cell invasion was performed using MCF-7 and MDA-MB-231 cells in cultures that contained TAM and TRAN alone or in combination and vehicle control. Briefly, 2×105 cells/well was seeded on 6-well plates for both cell lines and allowed to achieved confluence. A 1-mm width linear wound was created across the center of each well with a plastic tip. Wounded monolayers were then washed three times with medium to remove cell debris and incubated in the absence or 2 μM TAM, 200 μM TRAN and in combination both for 48 h. Photographs of the wounds were captured on day 0 and again 48 h later under phase contrast microscope. After photography, cells were incubated at 37 °C in a humidified incubator containing 5% CO2 in medium containing 2% serum in the absence or doses of drugs for 48 h and allowed to migrate. Experiments were repeated three times, in triplicate for both cell lines.
Migration assay
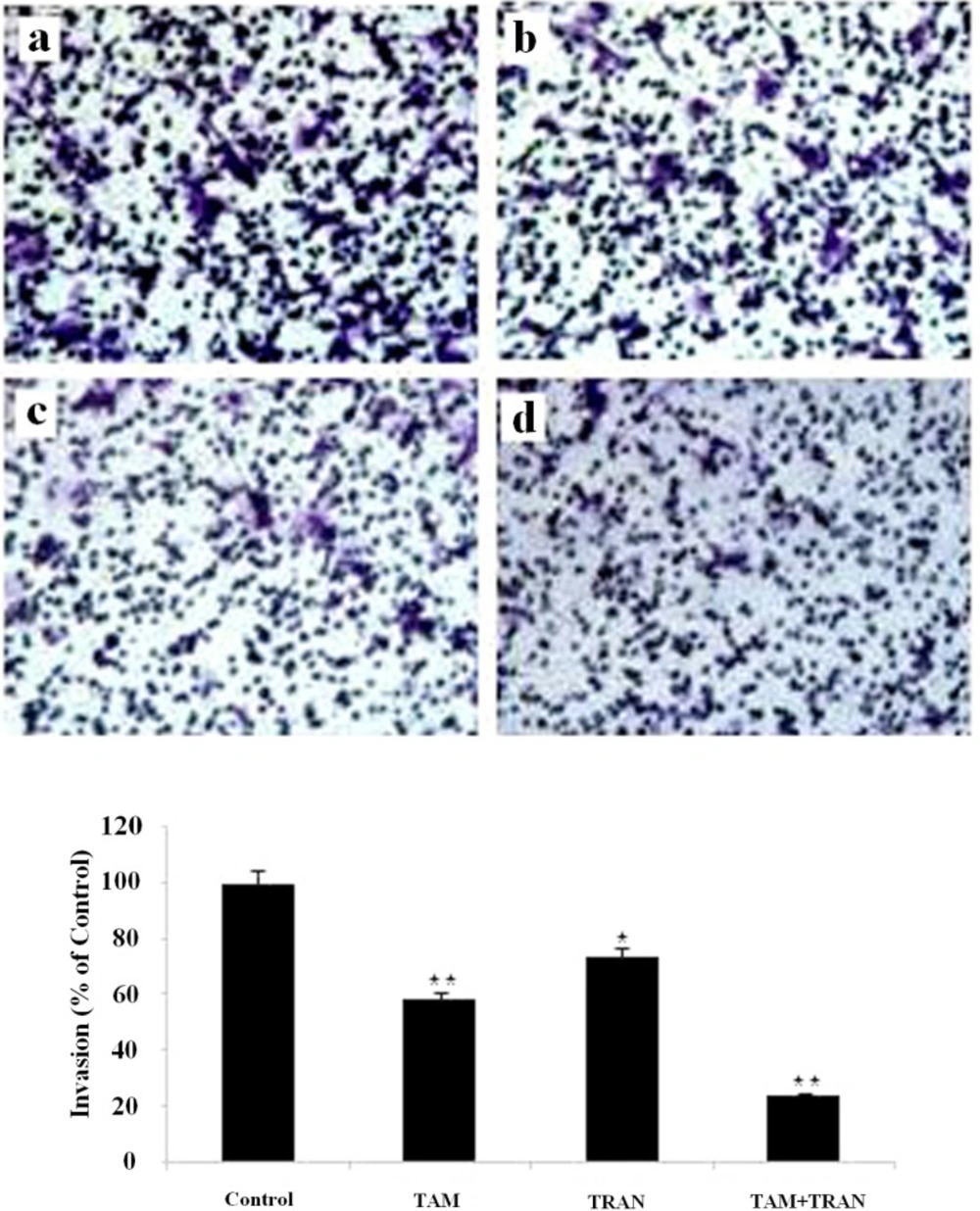
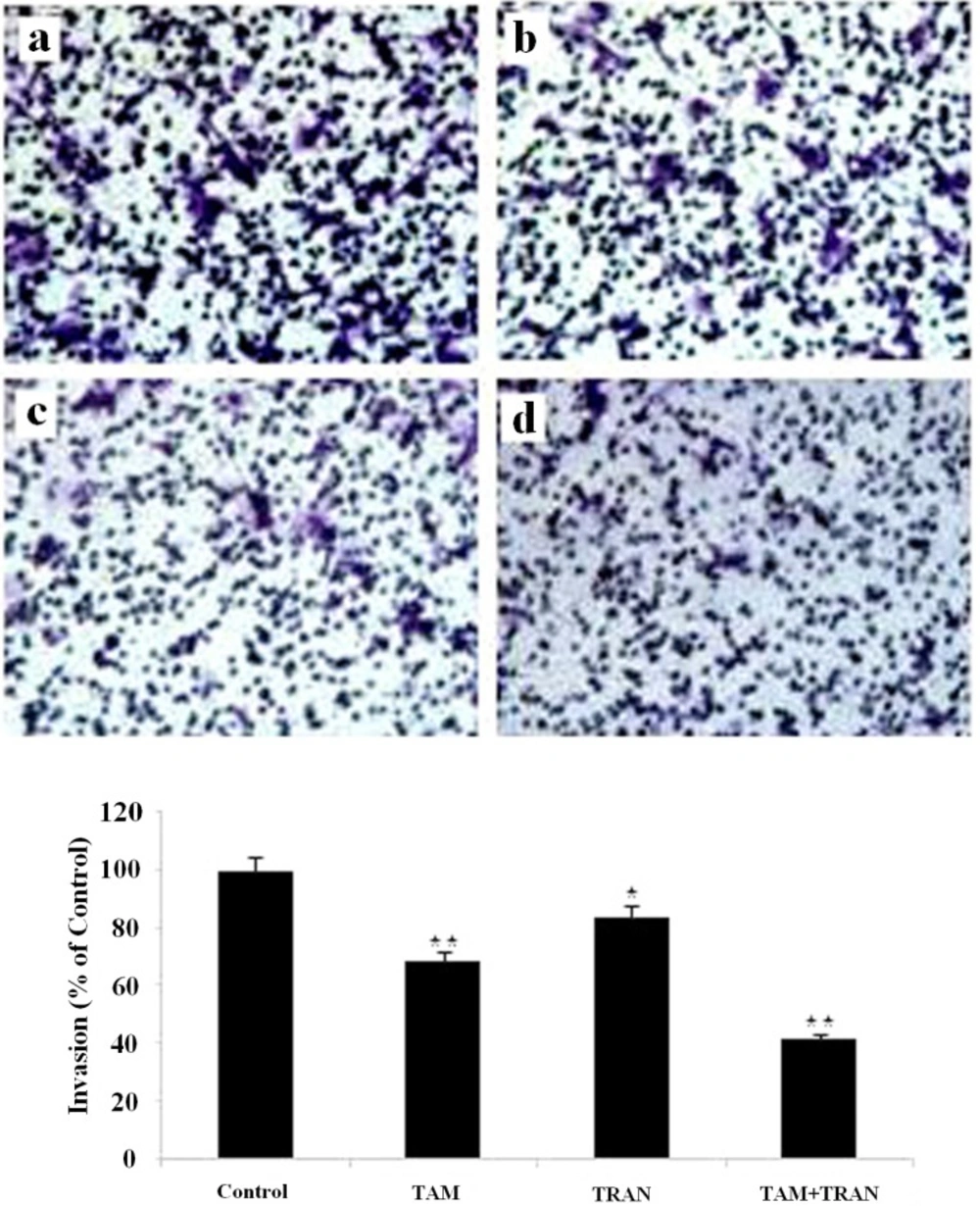
Cell invasion assay were performed by transwell chambers with polycarbonate membrane filters (8 µm pore membrane).
Transwell filters in 6-well plates were coated with matrigel, hydrated for at least 2 h in the tissue culture incubator with 500 μL serum free RPMI in the bottom and 500 μL in the top of the chamber. 5×105 MCF-7 or MDA-MBA-231 cells were plated in 500 μL serum-free RPMI on top of chamber, while 2 mL RPMI 10% FBS were placed in the lower chambers. Drugs (2 μM TAM, 200 μM TRAN or combination both) were added to the upper chambers. Cells without any drug were used as control. After Forty eight hours of incubation, the filters were removed, washed twice in PBS and fixed in 10% formalin for 15 min. After fixing at room temperature, the chambers were rinsed in PBS and stained with 0.2% crystal violet for 30 min. After washing the chambers by PBS, the cells at the top of the matrigel membrane were removed by several Q-tips.
Now all cells that remain are the ones that have invaded and made it to the bottom side of the membrane. The number of cells was counted in 10 randomly chosen fields using an inverted microscope and plotted as the percentage of invading cells of the total number of the cells. Three independent experiments each one in triplicate was carried out for both cell lines.
ELISA assay
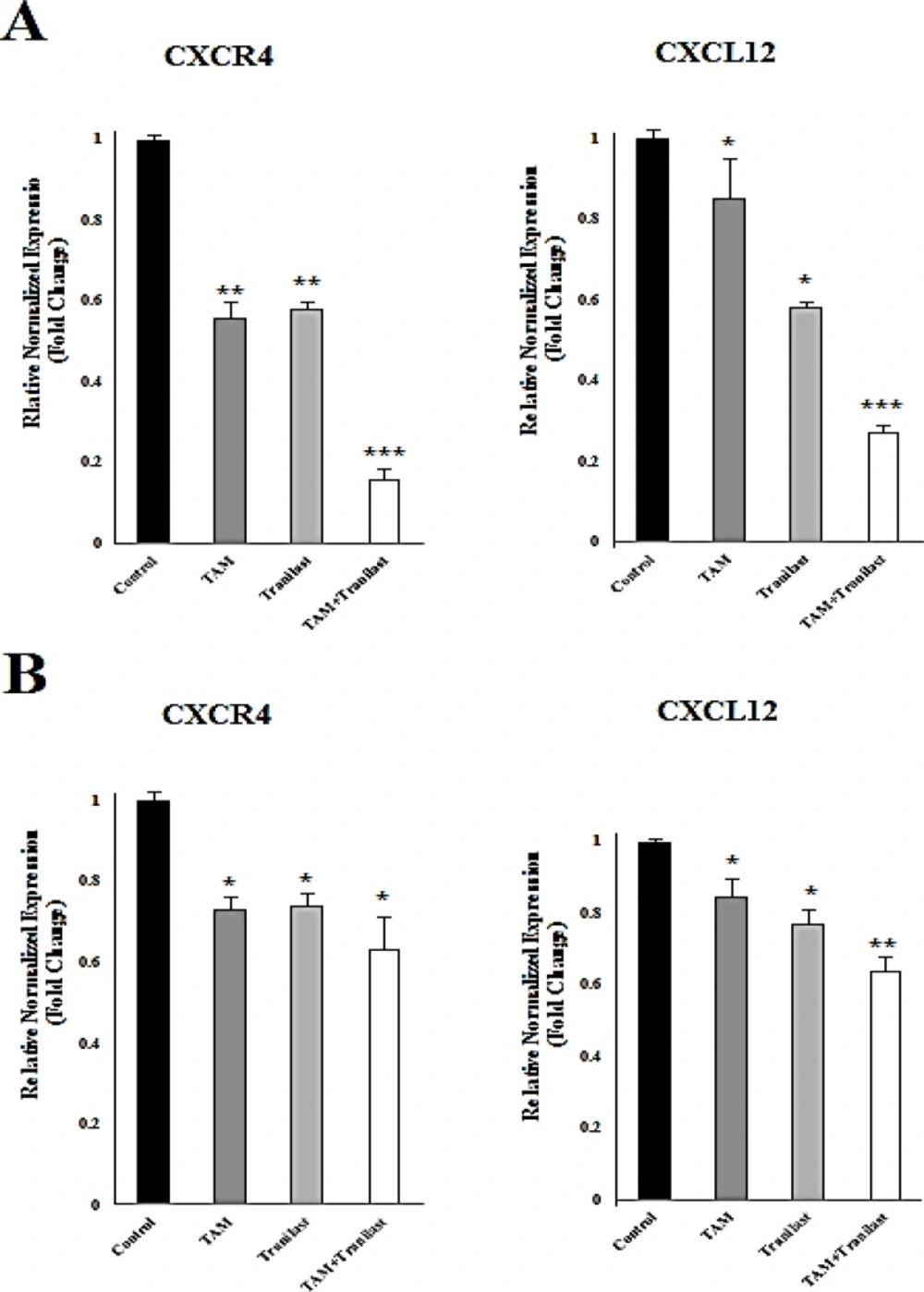
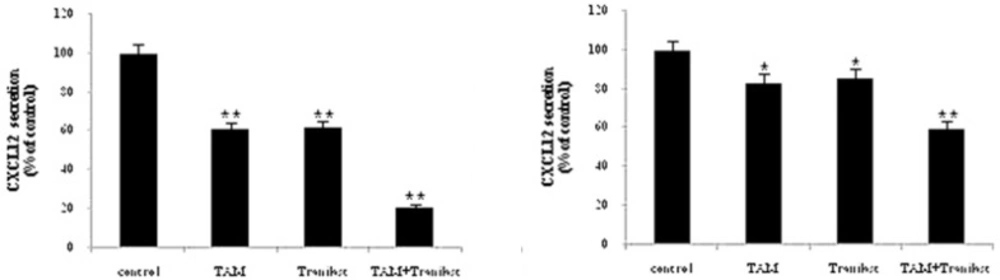
ELISA assay were used to determine the CXCL12 protein levels in MCF-7 and MDA-231 cells. Each of the both cell lines (5×104 cells/well) were plated in 24-well plates in 300 µL of medium. Cell culture supernatants were collected after 48 h of treatment with TAM (2 µM) and TRAN (200 µM) or combination of both drugs. CXCL12 concentration was determined using the Quantikine kit (R and D Systems) according to the manufacturer's instructions.
Quantitative gene expression analysis of CXCL12 and CXCR4
For quantitative real-time RT-PCR, total RNA was isolated from each cell line at 48 h after treatment with 2 µM TAM and 200 µM TRAN or combination both by using RNeasy Plus Mini kit (Qiagen). First-strand cDNA synthesis and amplification were done using QuantiTect Reverse Transcription Kit (Qiagen). Real time RT-PCR were performed using human-specific primers to CXCL12 forward: ccatgccgattcttcgaaag; reverse: ttcagccgggctacaatctg (product size: 101 bp), CXCR-4 forward: gccttatcctgcctggtattgtc; reverse: gcgaagaaagccaggatgaggat (product size: 130 bp) and GAPDH forward: actctggtaaagtggatattgttgc; reverse: ggaagatggtgatgggatttc (product size: 162 bp), with QuantiFast® SYBER® Green PCR Master Mix (Qiagen) on an iCycler with a multicolor real-time PCR detection system (Bio-Rad,Hercules, CA). The quantity of CXCR4 and CXCL12 transcripts was standardized by human glyceraldehyde-3-phosphate dehydrogenase (GAPDH). All PCRs were performed in triplicate.
Statistical analysis
All assays were performed in triplicate in three independent and separate experiments. For all experiments we used SPSS version17. Statistical analysis was performed using the one-way ANOVA followed by turkey's-test. p-values less than 0.05 were considered statistically significant.