Reagents and chemicals
The fluorescent probe propidium iodide (PI), and Triton X-100 were purchased from Sigma (St Louis, MO, USA), MTS from Promega (Madison, WI, USA), RNase A and Proteinase K from Fermentas (Vilnius, Lithuania); RPMI-1640 and FCS from Gibco (Grand Island, USA).
Plant materials
Roots of S. pachycarpa were collected in June 2010 at the Ferdowsi University Campus, Mashhad, Razavi Khorasan province, northeast of Iran.
The plant was identified by Mr. M. R. Joharchi from Ferdowsi University of Mashhad Herbarium (FUMH). A voucher specimen (No. 06-019-016) is deposited in the herbarium of the School of Pharmacy, Mashhad University of Medical Sciences, Mashhad, Iran.
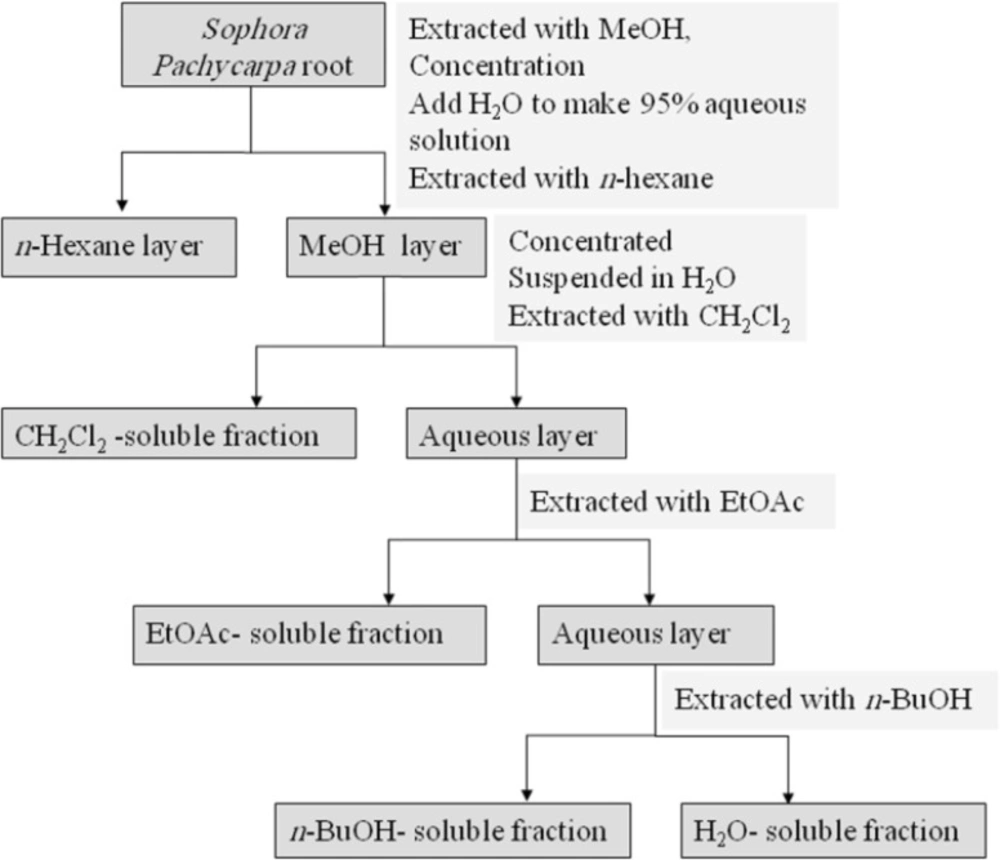
The dried root (380 g) was percolated with methanol (MeOH) at room temperature. The whole extract was filtered and the solvent was evaporated under reduced pressure at 40-45°C, to afford crude methanol extract. Methanol extract was then resolved in MeOH: H
2O 95:5 and partitioned successively between
n-hexan, dichloromethane (CH
2Cl
2), ethylacetate (EtOAc),
n-butanol (
n-BuOH), and finally H
2O. CH
2Cl
2, EtOAc, and
n-BuOH fractions were evaporated under vacuum and H
2O fraction was freeze dried to 0.4, 5.2, 3.5, and 5.9 g north of respectively. Fractions were stored at 4°C until analysis. A partitioning scheme of
S. pachycarpa methanol extract is presented in
Figure 1 (
27).
Partitioning scheme using immiscible solvents
Sample preparation
To prepare the stock solutions (100 mg/mL), each extract was dissolved in DMSO. The concentrations of 3.5-250 µg/mL were then obtained by diluting these solutions with Roswell Park Memorial Institute-1640 (RPMI-1640) so that the final concentrations of DMSO did not exceed 0.25%. All dilutions were prepared fresh before addition to the cells.
Cell culture and Treatment
A549, HeLa, HL-60, MCF-7, and PC3 cells were obtained from Pasteur Institute (Tehran, Iran) and maintained at 37ºC in a humidified atmosphere (90%) containing 5% CO2. Cell lines were cultured in RPMI 1640 supplemented with 10% (v/v) fetal bovine serum, 100 U/mL penicillin and 100 mg/mL streptomycin. Cells were seeded overnight and then incubated with various concentrations of different extracts for 48 h.
For MTS assay, cells were seeded at 104 cell per well onto 96-well culture plates. For assay of apoptosis, cells were seeded at 105 cell per well onto a 24-well plate.
For each concentration and time course study, there was a control sample which remained untreated and received an equal volume of the solvent. Paclitaxel (
41) was used as positive control (350 nM).
Cell viability
The MTS assay (
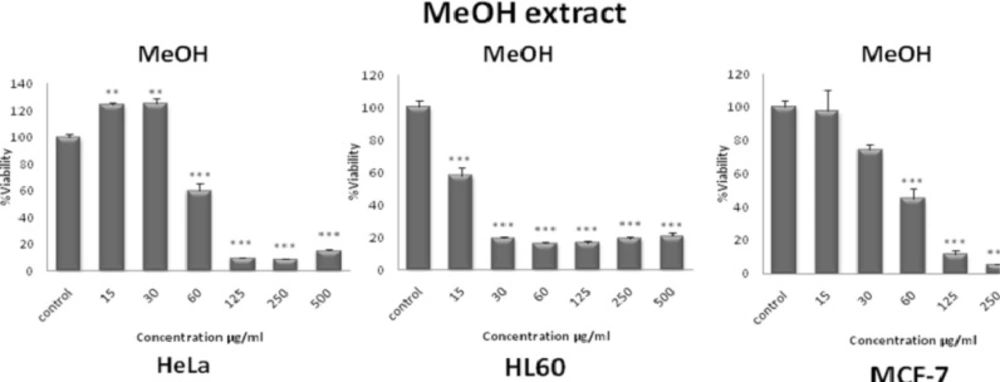
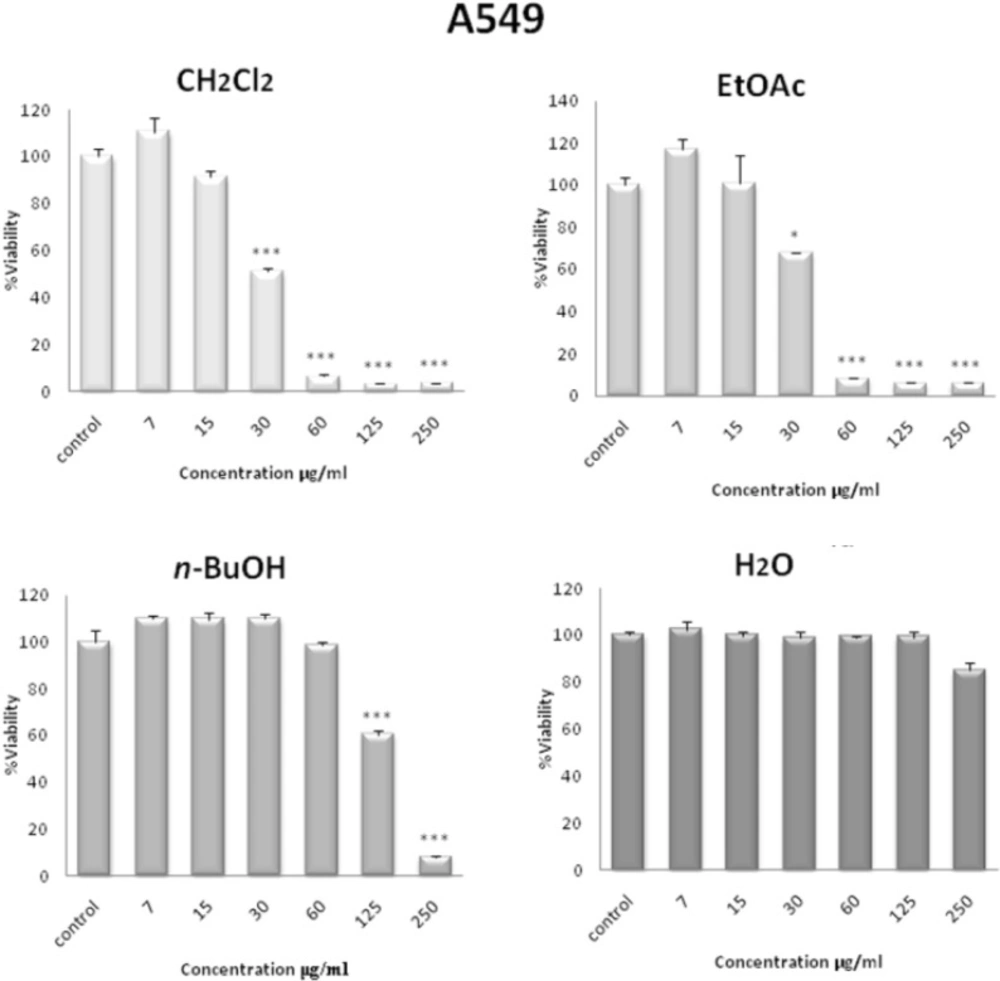
42) is based on the reduction of MTS by mitochondrial dehydrogenases in metabolically active cells, to form the colored, water-soluble formazan. Cells were seeded in each well of a 96-microwell plate and treated with various concentrations of methanol extract and different fractions obtained from
S. pachycarpa root. After incubation for 48 h, CellTiter 96® Aqueous One Solution Reagent (Promega, Madison, WI, USA), which is composed of the novel tetrazolium compound MTS and an electron coupling reagent phenazine methosulfate (PES, a redox intermediary), was added to each well according to the manufacturer’s instructions. After 1 h, the cell viability was determined by measuring the absorbance at 490 nm using an ELISA microplate reader (Awareness, Palm City, FL, USA).
Cytotoxicity was expressed as IC50, which was calculated using Graph Pad prism 5 software and presented as mean ± SEM of three independent experiments with three replicates for each concentration.
Paclitaxel was used as a positive control.
Leukocyte culture
Human umbilical cord blood samples (50 ml) were collected from a fresh umbilical cord attached to the placenta by gravity flow in sterile 50 ml syringe containing citrate buffer as an anticoagulant. The sample was diluted with an equal volume of Phosphate Buffered Saline (PBS), layered over Ficoll-Hypaque solution according to density gradient (1.077 g/mL), and centrifuged at 800 g for 20 min at room temperature. The mononuclear cell layer was removed, washed twice in PBS and resuspended in RPMI 1640 medium supplemented with 10% (v/v) fetal bovine serum, 100 U/mL penicillin and 100 mg/mL streptomycin. Leukocytes (5×104 cells per well) were treated with different concentrations of each fraction of S. pachycarpa in 96-well plates, for 48 h. This study protocol was approved by the ethical committee of Mashhad University of Medical Sciences.
Apoptosis
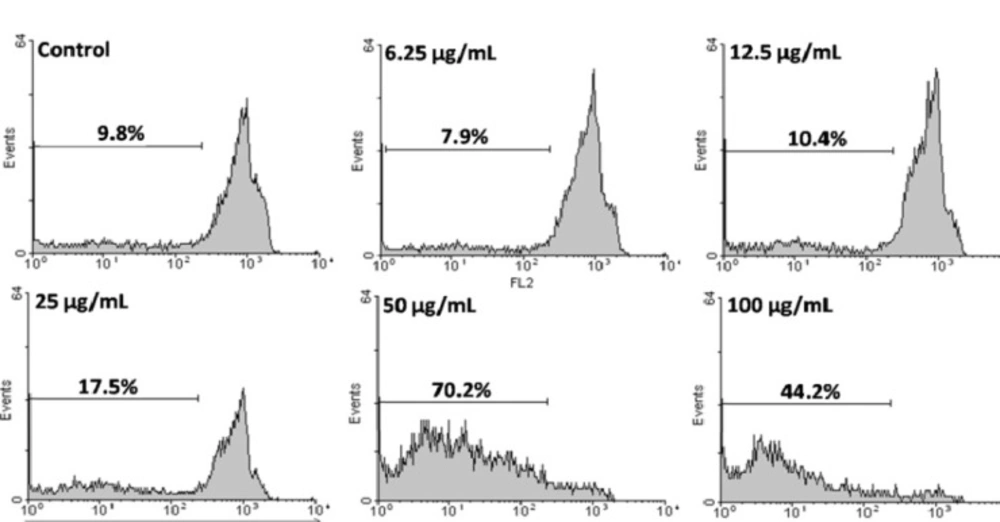
Apoptotic cells were detected using PI staining of treated cells followed by flow cytometry to detect a sub-G1 peak (
43,
44).
It has been reported that DNA fragmentation creates small fragments of DNA that can be eluted following incubation in a hypotonic phosphate citrate buffer. When stained with a quantitative DNA-binding dye such as PI, cells that have lost DNA will take up less stain and will appear to the left of the G1 peak. Briefly, HeLa cells were cultured overnight in a 24-well plate and treated with various concentrations of CH2Cl2 extract for 48 h. Floating cells were harvested and incubated at 4°C overnight in the dark with 750 µL of a hypotonic buffer (50 µg/mL PI in 0.1% sodium citrate + 0.1% Triton X-100) before flow cytometric analysis using a Partec flow cytometer (GmbH, Münster, Germany) was conducted. Ten thousand events were acquired.
Statistics
One-way analysis of variance (ANOVA) and Bonferroni’s post hoc were used for data analysis. All results were expressed as mean ± SEM and p-values <0.05 were considered statistically significant.