
Introduction
Fudosteine, (-)-(R)-2-amino-3-(3-hydroxypropylthio) propionic acid (Figure 1), is a new cysteine derivative, approved in Japan in 2001 as a new muco-active agent with indications for chronic respiratory diseases such as bronchial asthma, chronic bronchitis, pulmonary emphysema, bronchiectasia, pulmonary tuberculosis, pneumonoconiosis, atypical mycobacterial disease and diffuse panbronchiolitis (1-7).
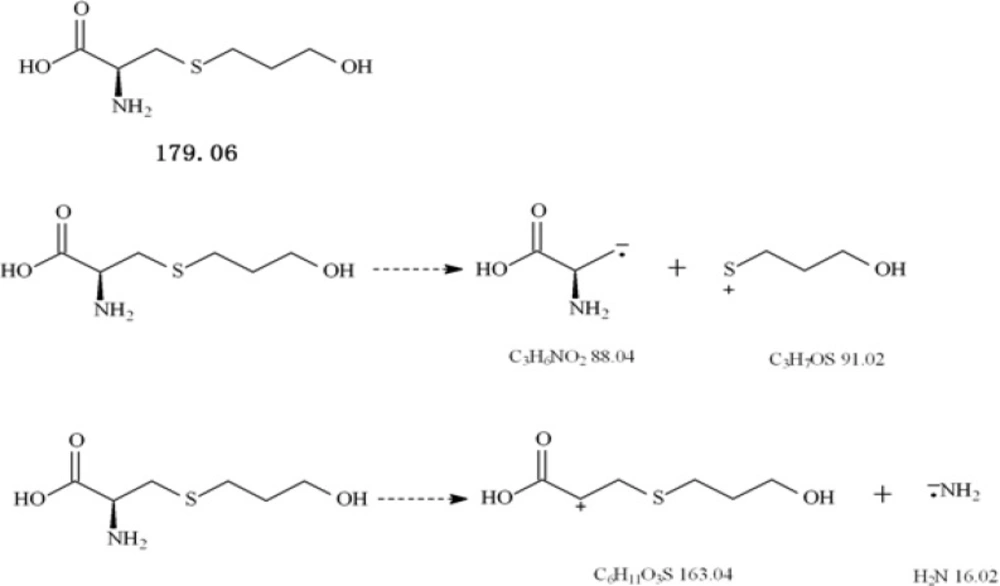
Figure 1
Chemical structures of fudosteine and the probable mass spectral fragmentation pathways of fudosteine
Up to now only a few analytical methods has been reported for the determination of fudosteine in biological samples (8-11). As other amino acid, fudosteine was very polar, and lacked UV absorption or fluorescent functional groups. UV and fluorescence detections are not sufficiently selective for the determination of fudosteine in human plasma as the interference of the endogenous amino acids. Quantitation of fudosteine was challenging due to the lack of significant UV absorption and low levels of the drug in biological fluids. One of the existed analysis method was employed precolumn derivatization for sensitive detection, which was time consuming and increase variation (9). Another reported method chose CN column (11). In our study we used ODS column which was more common to fulfill the assay.
In this paper we developed a novel approach to determine fudosteine, in order to allow it using as a high throughput tool during a pharmacokinetic assessment of fudosteine.
Experimental
Reagent and chemicals
Fudosteine reference (99.0% purity) and capsule formulation of fudosteine 200 mg (Batch no. 060901) were all from Dilong Pharmaceutical (Group) Co., Ltd. (Helongjiang, PR China). HPLC-grade LiChrosolv acetonitrile was from Merck (Darmstadt, Germany). Acetic acid was from DIMA (DIMA, USA). Aqua pro injection (Changfu, Shandong, China) was used as solution water. All other chemicals were of analytical grade. The blank human serum was obtained from the Metachysis Centre of Lanzhou General Hospital (Lanzhou, Gansu, China). Pre-analyzed blood samples were from health volunteers. Samples were identified with a coded number and were completely anonymous.
HPLC-ESI/MS/MS apparatus and conditions
HPLC was performed using a Shimadzu LC-20A Series (Shimadzu, Kyoto, Japan), which include LC-20AD binary pump, DGU-20A3 degasser, CBM-20A communications bus module, SIL-20AHT auto sampler, CTO-20A column oven. HPLC separations were carried out on a Rainin Microsorb-MV ODS column (4 μm, 150 mm×4.6 mm i.d.) from (Emeryville, CA, USA). Separations were performed at 30 ℃ temperature. The mobile phase composition was composed of acetonitril and 20 mM acetic acid (25:75, v/v), which was pumped at a flow rate of 0.40 mL/min. The overall chromatographic run time was approximately 7 min. The autosampler was set with an injection volume of 10 μL.
Mass spectrometric detection was performed on an API 3200 triple quadrupole instrument (ABI-SCIEX, Toronto, Canada) using Multi-Reaction Monitor (MRM). A turbo electrospray interface in positive ionization mode was used. The main working parameters of the mass spectrometer are summarized in Table 1. Data acquisition and processing was performed on Analyst 1.4.2 software package (SCIEX).
Table 1MS parameters for monitoring fudosteine
| Parameter | Value |
|---|---|
| Source temperature (◦C) | 500 |
| Dwell time per transition (ms) | 200 |
| Ion source gas (Gas 1) (psi) | 60 |
| Ion source gas (Gas 2) (psi) | 70 |
| Curtain gas (psi) | 20 |
| Collision gas (psi) | 5 |
| Ion spray voltage (V) | 5500 |
| Declustering potential (DP) (V) | 16 |
| Entrance potential (V) | 10 |
| Collision energy (V) | 22 |
| Collision cell exit potential (V) | 14.7 |
| Mode of analysis | Positive |
| Ion transition for fudosteine, m/z | 180.2/91.0 |
Clinical study design
The phase I clinical pharmacokinetic study was conducted with the approval of the Committee of Ethics of clinical study of the General Hospital of Lanzhou. Five male and five female Chinese volunteers participated this phase I clinical pharmacokinetic study. Single dose oral administration study: Each subject received a single oral dose of fudosteine capsule of 200 mg. Blood samples (3 mL) for assay of serum concentration of fudosteine were collected at times of 0, 0.5, 0.75, 1, 1.5, 2.0, 3.0, 4.0, 6.0, 8.0, 12, 24, 36 and 48 h after oral administration of fudosteine. They were put into centrifuge tubes and were centrifuged immediately at 2000 g for 10 min to obtain serum for assay of fudosteine, which was then frozen at -40 °C until analyzed.
Sample preparation
Human serum samples were obtained from the Blood Transfusion Center of the General Hospital of Lanzhou (Lanzhou, Gansu, China) and were stored under -40 ℃. Before use, the serum samples were thawed at 37 ℃ water bath and centrifuged at 13000 g for 10 min at ambient temperature. Aliquots were spiked with the diluted standard solutions and these serum samples were prepared daily.
Bioanalytical method validation
The method was validated prior to the analyses of plasma samples according to the guidance of bioanalytical method validation. The limit of quantification, specificity, linearity, accuracy, precision and stability of fudosteine in plasma sample were assessed and evaluated.
Preparation of stock solutions
Stock solutions of fudosteine were prepared at 1 mg/mL in water and stored at -40 ℃ until use. Drug-free serum was used for validation studies. Working solutions for calibration and controls were prepared daily in water by appropriate dilution at 1.0, 10.0 and 100.0 μg/mL.
Calibration curve and control samples
Calibration curves were prepared by spiking different samples of 0.5 mL blank serum each with an appropriate volume of one of the above-mentioned working solutions to produce the calibration curve points equivalent to 0.01, 0.10, 0.45, 1.0, 4.5, 10.0, 15.0 μg/mL of fudosteine.
Standard curves were prepared daily and constructed by linear regression analysis of the fudosteine peak area ratio versus the respective concentration of fudosteine. Another stock solution of fudosteine was separately prepared at six different concentrations i.e. 0.10, 0.45, 1.0, 4.5, 10.0, 15.0 μg/mL for quality control (QC).
Accuracy and precision
The accuracy and precision were assessed by determining the quality control samples at six concentration levels of fudosteine on 3 consecutive days. Precision was expressed as relative standard deviation (R.S.D.) and accuracy as [(mean found concentration - added concentration)/(added concentration)]×100%. Intra-day precision and accuracy were determined by repeated analysis of the QC samples on one day (n = 5), while inter-day precision and accuracy by repeated analysis was conducted for 3 consecutive days (n = 5 series per day).
Stability
Drug stability in a biological fluid is a function of the storage conditions, the chemical properties of the drug, the matrix and the container system. The analyte was stable during sample collection and handling, after short-, long-term storage, after going through freeze and thaw cycles and during the analytical process. Conditions used in stability experiments reflected situations likely to be encountered during actual sample handling and analysis. The stability of analyte in stock solution was also proven.
Results and Discussion
MS specificity and selectivity
The MS tuning and MS/MS dissociation study were optimized for standard compound by varying the cone voltage and collision energy. The flow injection analysis mode was used to optimize the MS conditions. Working solutions were directly injected to the electrospray probe at a flow rate of 0.005 mL/min. The Q1 scan picture is shown in Figure 2. The MS2 scan experiments displayed daughter scan spectra of fudosteine. The most abundant ions from daughter ion experiments were selected to obtain MRM transitions as more sensitive as possible. Positive and negative ion modes were tried for fudosteine, and the results suggested that the positive ion mode was more sensitive, so positive ion mode was chosen. The probable mass Spectral Fragmentation Pathways of fudosteine were shown in Figure 1.
The ESI source gave [M+H]+ ions as quasimolecular ions afforded a diagnostic ion at m/z 179.8. The MS2 scan experiments displayed the daughter scan spectra of fudosteine. The [M-H]+ ions were selected as the precursor ions to perform the MS/MS experiments, and multiple product ions were observed. The main daughter ions of fudosteine were m/z 163.0 and m/z 91.2 (Figure 2), due to there was an interference in blank serum at m/z 163.0 we finally chose m/z 179.8/91.2 as the quantification daughter ion to obtain the MRM transitions as sensitive as possible.
Blank serum after sample preparation was monitored as total ion chromatogram did not show any peak. Specific fudosteine ion chromatograms showed just one peak at relative retention time of 3.60 min respectively.
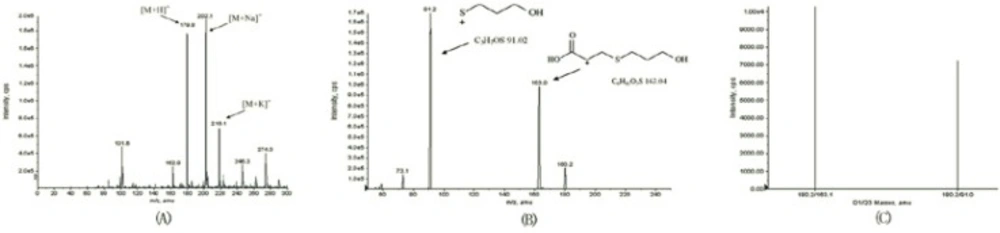
Figure 2
The Q1 and the MS2 scan pictures
Limit of quantification and limit of detection
As the accuracy profile is comprised within the acceptance limits, the LOQ was fixed to 100 ng/mL, i.e. the smallest concentration level investigated. Indeed, precision and trueness were demonstrated at this concentration level (Table 1). The LOD has been estimated at 10 ng/mL (S/N=10:1). And analyte peak was identifiable, discrete and reproducible with a precision of 20% and on accuracy of 80–120%. Figure 3 shows typical chromatograms obtained from samples of blank serum sample (m/z 180.1/91.0 ) (A), serum spiked with 2.1 μg/mL of fudosteine(m/z 180.1/91.0) (B) and fudosteine in serum of healthy volunteer an hour after a single oral dose of 200 mg fudosteine (C). Visual examination of the HPLC-ESI/MS/MS chromatograms of blank samples obtained during the validation, including the serum from six volunteer s, indicated high specificity. The analytes were chromatographically resolved and no significant interferences from endogenous material were observed.
The mean calibration equation was y=109.07x-4.69, γ=0.9998, where y represents the plotting peak areas of the analyte and x represents the plasma concentration of fudosteine in μg/mL. Calibration curves showed an excellent linearity at the range 0.1-15.0 µg/mL.
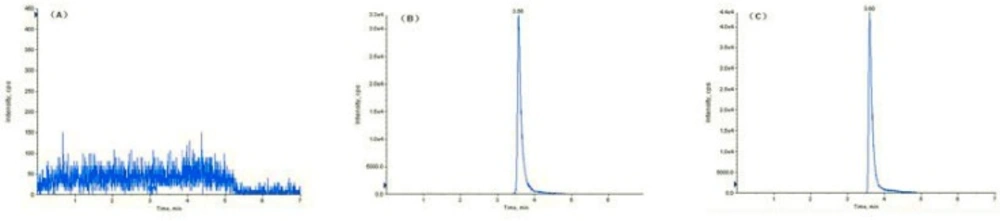
Figure 3
Mass spectrometry of (A) blank serum sample 180.1/91.0, (B) serum spiked with 2.1 μg/mL of fudosteine, 180.1/91.0 (C) fudosteine in serum of healthy volunteer an hour after a single oral dose of 200 mg fudosteine
Precision and accurate
Table 2 showed the results of inter- and intra-day precision and accuracy of fudosteine. All the results were within the specified ranges and therefore acceptable.
The R.S.D. values presented in Table 2 were low. The relative standard deviation values for inter-day and intra-day precision were within 2.0% and 5.0%, illustrating the good precision of the proposed method.
Table 2Precision, accuracy and recovery for determination of fudosteine in spiked serum (n=5).
| Concentration (ng/mL) | Precision (R.S.D.%) | Accuracy (%) | |
|---|---|---|---|
| Within-day | Between-day | ± s | |
| 100 | 1.6 | 4.8 | 109.9±5.1 |
| 450 | 0.4 | 1.3 | 110.4±1.4 |
| 1000 | 0.7 | 1.6 | 100.1±1.6 |
| 4500 | 1.2 | 3.6 | 95.8±1.1 |
| 10000 | 1.6 | 1.6 | 100.7±1.6 |
| 15000 | 1.1 | 4.5 | 99.8±1.5 |
Stability
The analyte was found to be stable and the range of accuracy was from 95.8 to 110.4%. Fudosteine was shown to be stable in serum at room temperature for at least 2 h and the post preparative samples were stable in autosampler for at least 8 h. Fudosteine had an acceptable stability at −20 ℃ for 1 month.
Application of the method
This method had been successful applied to the analysis of healthy volunteer serum samples proving the advantage of the method developed in this study. Figure 4 showed a serum concentration curve obtained during the phase I clinical pharmacokinetic study. The serum concentration of fudosteine was found from 0.11 to 9.5 µg/mL.
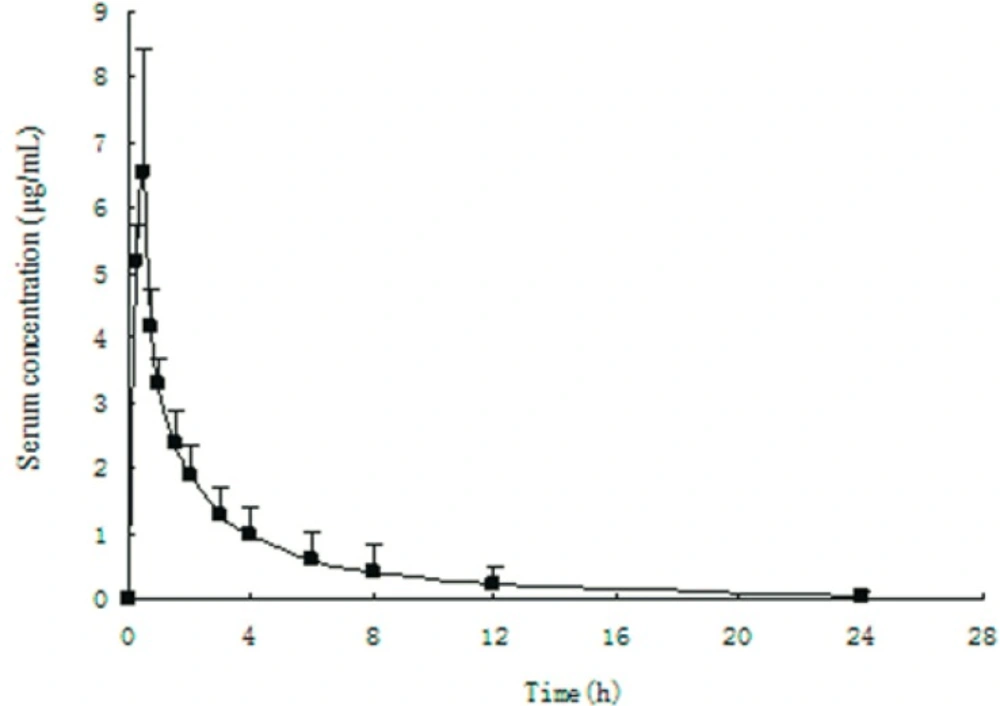
Figure 4
Mean serum concentration-time curve in 10 healthy volunteers when administered oral doses of 200 mg fudosteine during the phaseⅠclinical pharmacokinetic study
Comparing to the reported method
There are only a few reports on assaying fudosteine in blood. Xu et al. reported an assay method for fudosteine in plasma employing pre-column derivatization which had to go through a time-consuming and arduous pretreatment step (9). The comparison of current method and previous methods was in Table 3. HPLC-MS/MS method had saved a lot of preparation time as it was unnecessary to perform pre-column derivatization. Unlike previous HPLC-MS/MS analysis method (11) in our work we chose positive mass analysis mode which got good ionization efficiency. We selected MRM mode for the mass spectrometry. The ion transition for fudosteine was m/z 180.2/91.0 (Figure 2). Wen et al. reported that CN columns but not ODS columns was suitable for the determination of fudosteine as the later had almost no retention (11). In our study we found it was possible to use common ODS column for the quantification of fudosteine while using acetonitril and 20 mM acetic acid (25:75, v/v) as mobile phase. Under this condition the efficiency of ionization was good in positive mass scan mode and the ion suppression was not obvious. The retention time of fudosteine was 3.6 min with good peak shape (Figure 3). We used ODS column to get similar precision and accuracy as Wen et al. Considering the results above, the submitted method can be used as a simpler procedure with similar precision and accuracy but with few pretreatment procedure and analysis time consuming.
Table 3The comparison of current method and previous methods
| Analytical method | Analytical column | Precision | Accuracy | The calibration curve | γ | Precolumn derivatization | Total test time |
|---|---|---|---|---|---|---|---|
| HPLC (8) | ODS | <7.3% | 96.8-106.6% | 0.3-40.0 μg/mL | 0.9998 | Yes | >18 min |
| LC-MS (9,10) | ODS | <10.67% | 93.71-99.51% | 0.05-20 μg/ml | 0.9977 | Yes | >28 min |
| LC-MS/MS (11) | CN | <9.2% | 95.7-107.5% | 0.02-10.0 μg/mL | >0.999 | No | <5 min |
| LC-MS/MS | ODS | <4.8% | 95.8-110.4% | 0.1-15.0 µg/mL | 0.9998 | No | <6 min |
Conclusions
A quantitative analysis method for fudosteine in human serum by HPLC-ESI/MS/MS was established, which showed high sensitivity and selectivity. The method was very simple and sample preparation was minimal. This method was successfully applied to study the pharmacokinetics of fudosteine in healthy Chinese volunteers. This assay provided a new strategy and approach to determine amino acid derivatives in biological samples.
