Compounds
OMT was purchased from Shanxi Huike Botanical Development Company Limited (Shanxi, China). OMT was dissolved in dimethyl sulfoxide (Sigma, MO, USA) to yield a 10 mg/mL stock solution and was diluted to the indicated concentrations in the experiments.
Cell culture
BV2, a murine microglial cell line, which is a suitable model for in-vitro study of microglia, was used in this study. The cells were grown in a flask (75 cm2) and washed with phosphate buffered saline (PBS) solution twice and then treated with trypsin-EDTA (TE; Biowest, Nuaille, France) in PBS for 3 min at 37°. The TE was inactivated by equal volume of 1×fetal bovine serum (FBS; HyClone, Utah, USA). The culture was centrifuged at 1000 rpm at 4° for 5 min and the pellet was resuspended in 10 mL of Dulbecco’s Modified Eagle’s Medium (DMEM, Sigma, MO, USA) containing 10% FBS and 1% antibiotic antimycotic cocktail (Sigma, MO, USA). The cells were counted using a hematocytometer and approximately 2×106 cells were plated into each flask containing 10 mL of 10% FBS in DMEM and grown at 37° and 5% CO2 in an incubator. The cells were subcultured every 2-3 days. For experiments, the BV2 cells were maintained in DMEM without antibiotics or FBS for the required periods of treatment (Basic medium).
Treatment of cell culture
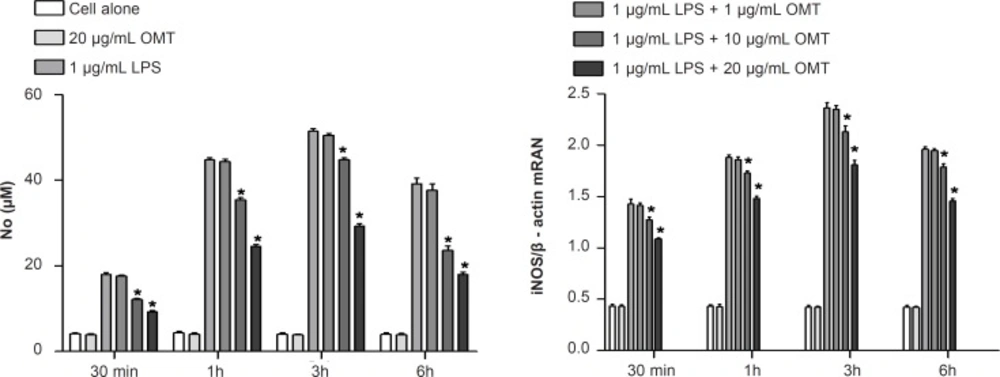
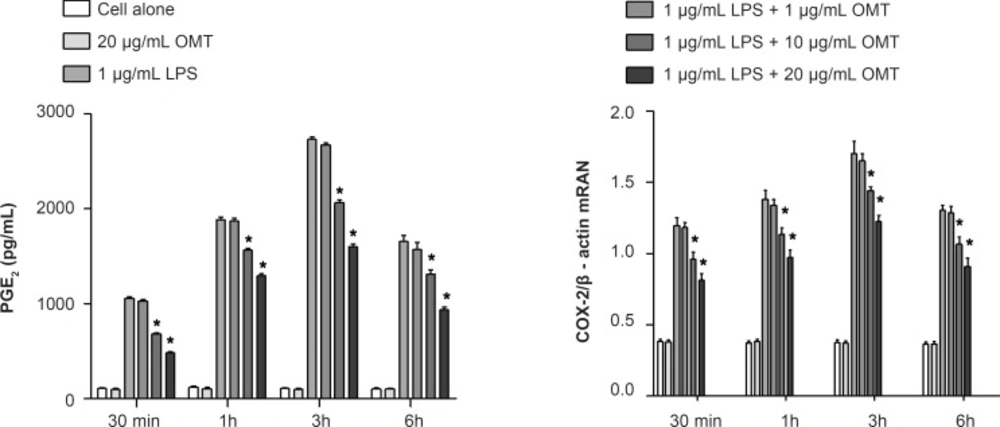
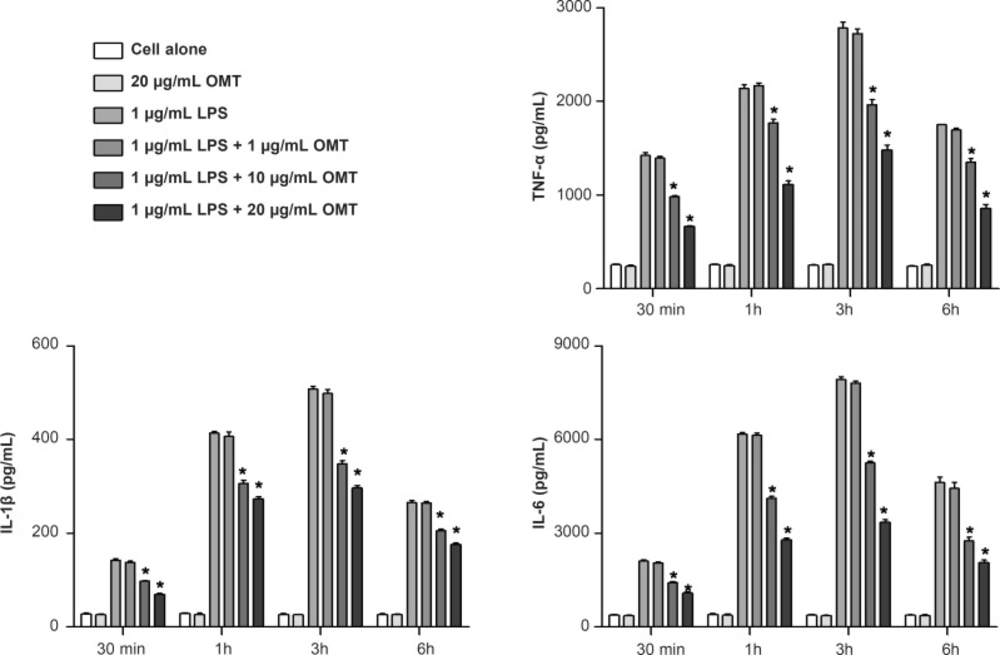
2×106 cells were plated onto cell culture dishes and grown in 10% DMEN/FBS with antibiotics overnight. On the following day, the cells were washed twice with PBS, transferred to basic medium and pretreated with different concentrations of OMT for 30 min as followed by stimulation with LPS (1 μg/mL; Sigma, MO, USA) for different times (30 min, 1 h, 3 h, and 6 h) in the incubator. The control was taken as cells grown in basic medium for the same time periods. The cells and supernatant collected were used for analysis. Each experiment was performed in three independent experiments.
Nitrite assays (Griess assay)
NO levels in the culture supernatants were measured by a Griess reaction. After cells (5×105 cells/mL) were stimulated in 24 wells for 24 h, 100 μL of each cultured medium was mixed with the same volume of the Griess reagent (1% sulfanilamide/0.1% N-(1-naphthyl)-ethylenediamine dihydrochloride/2.5% H3PO4). NO concentration was determined by measuring the absorbance at 540 nm with a Vmax 96-well microplate spectrophotometer. Nitrite concentration was calculated with reference to a standard curve of sodium nitrite generated by known concentrations. Results were expressed as μM.
Western blot analysis
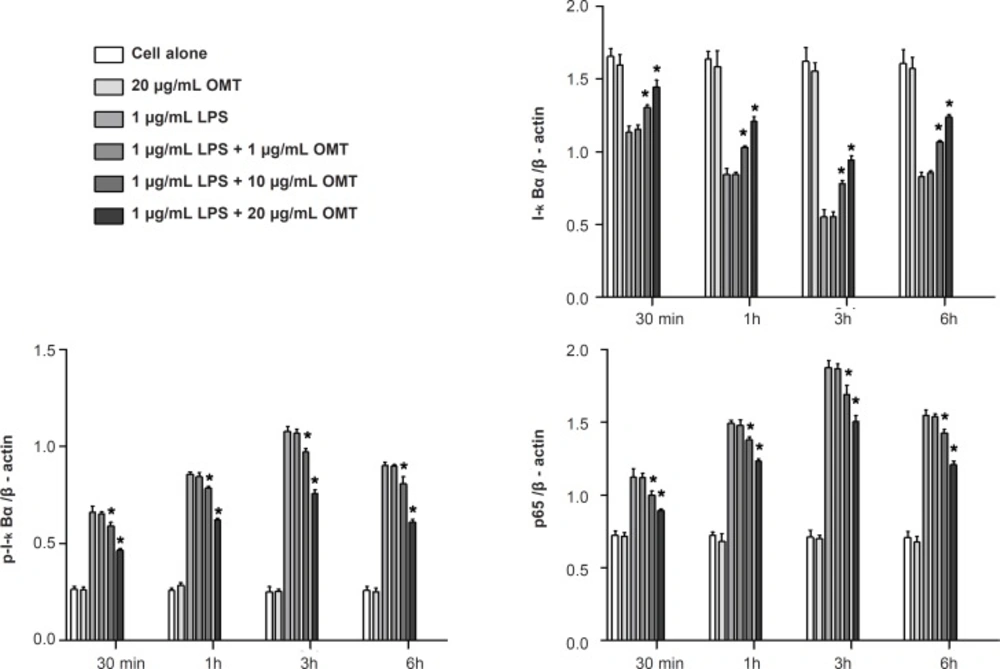
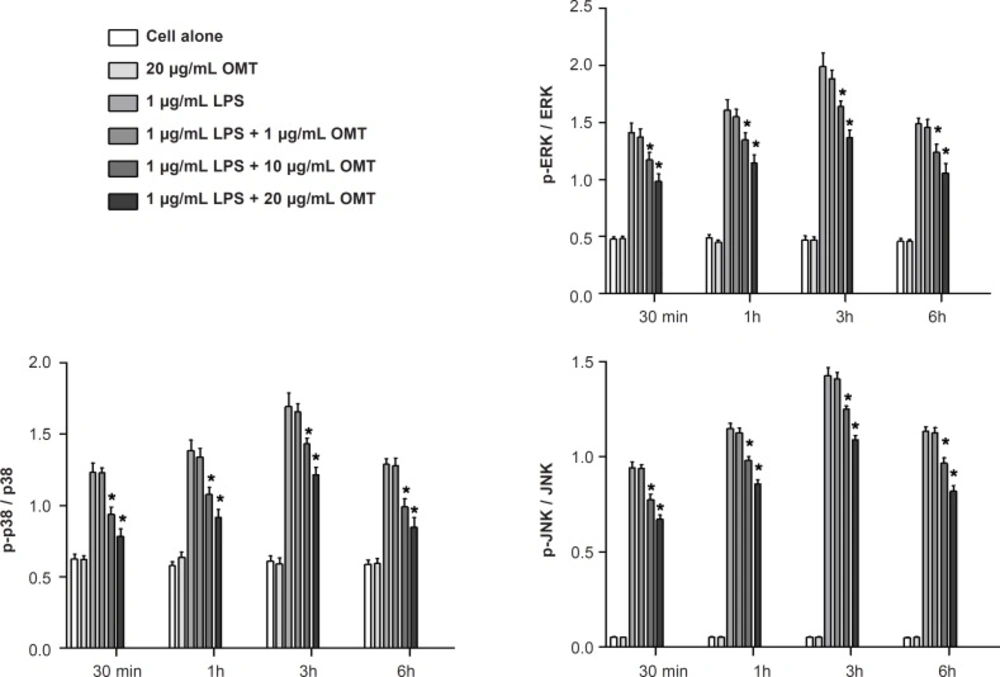
Cells were suspended in lysis buffer (1% Triton X-100, 1% deoxycholate, and 0.1% NaN3) and incubated for 30 min on ice. For nuclear extraction, cells were lysed with NE-PERTM (Pierce, Rockford, IL, USA). Protein concentrations were determined (Bio-Rad, Hemel, Hempstread,UK) and same quantity of proteins were resolved on 10% gels, and then transferred to nitrocellulose membranes (Millipore, Billerica, MA, USA). The membrane blots were blocked with 5% milk in TBS-T (o.1 M Tris-HCl, pH 7.4, 0.9% NaCl, 0.1% Tween-20) and incubated with primary antibodies against β-actin (Santa Cruz Biotech, Santa Cruz, CA), inhibitor of kappa B-alpha (I-κBα), phospho-I-κBα (p-I-κBα), p65 NF-κB, the phosphor (p) - or total forms of ERK 1/2, p38 MAPK and JNK (Cell Signaling Technology Inc., Beverly, MA, USA). The membranes were subsequently incubated with peroxidase-conjugated affinity goat anti-rabbit IgG (Sigma, MO, USA) and detected by chemiluminescence detection system (ECL, Amersham, Bershire, UK). Relative intensity was presented.
Enzyme-linked immunosorbent assay (ELISA)
The levels of TNF-α, IL-1β, IL-6 and PGE2 in the culture media were determined by ELISA. ELISA kits from R&D Systems (Minneapolis, MN) were employed for the measurement of TNF-α, IL-1β and IL-6, and a kit from Cayman Chemical (Ann Arbor, MI) was employed for the measurement of PGE2 in accordance with the manufactures’ instructions. Results were expressed as pg/mL.
Reverse transcription-polymerase chain reaction analysis (RT-PCR)
Total RNA was prepared from BV-2 cells by using the Trizol® reagent (Invitrogen Corporation, Carlsbad, CA, USA) according to the manufacturer’s protocol. RNA (2 μg) of each sample was used for synthesizing cDNA through inverse transcription; 1 μL of cDNA was used to carry out PCR amplification. Primers were synthesized by Shanghai Sangon Biological Engineering Technology Company Limited. Correctness of the gene order was proved in GenBank. Primers included 5’- CTGCAGCACTTGGATCAGGAACCTG-3’ (forward) and 5’-GGGAGTAGCCTGT GTGCACCTGGAA-3’ (reverse) for iNOS, 5’-TTGAAGACCAGGAGTACAGC-3’ (forward) and 5’-GGTACAGTTCCATGACATCG- 3’ (reverse) for COX-2, as well as 5’-AGCCATGTACGTAGCCATCC-3’ (forward) and 5’-GCTGTGGTGGTGA AGCTGTA -3’ (reverse) for β-actin. PCR amplification of the resulting cDNA template was conducted by using the following conditions for 45 (β-actin), 36 (COX-2) or 27 (iNOS) cycles. After an initial denaturation step at 95°C for 15 min, temperature cycling was initiated. Each cycle consisted of denaturation at 94°C for 15 sec, annealing at 60°C for 25 sec, and elongation at 72°C for 20 sec (β-actin). After an initial denaturation step at 95°C for 5 min, temperature cycling was initiated. Each cycle consisted of denaturation at 94°C for 30 sec, annealing at 57 °C for 45 sec, and elongation at 72°C for 30 sec (COX-2). After an initial denaturation step at 95°C for 5 min, temperature cycling was initiated. Each cycle consisted of denaturation at 94°C for 45 sec, annealing at 60°C for 45 sec, and elongation at 70°C for 1 min (iNOS). RT-PCR products (5 μL) were analyzed by 2% agarose gel electrophoresis. The gray scale of the electrophoresis strip was scanned by an ultraviolet photometry (UVP) gel imaging system. The relative expression of products was normalized to β-actin mRNA; data were analyzed with an image analysis system.
Cytotoxicity assay
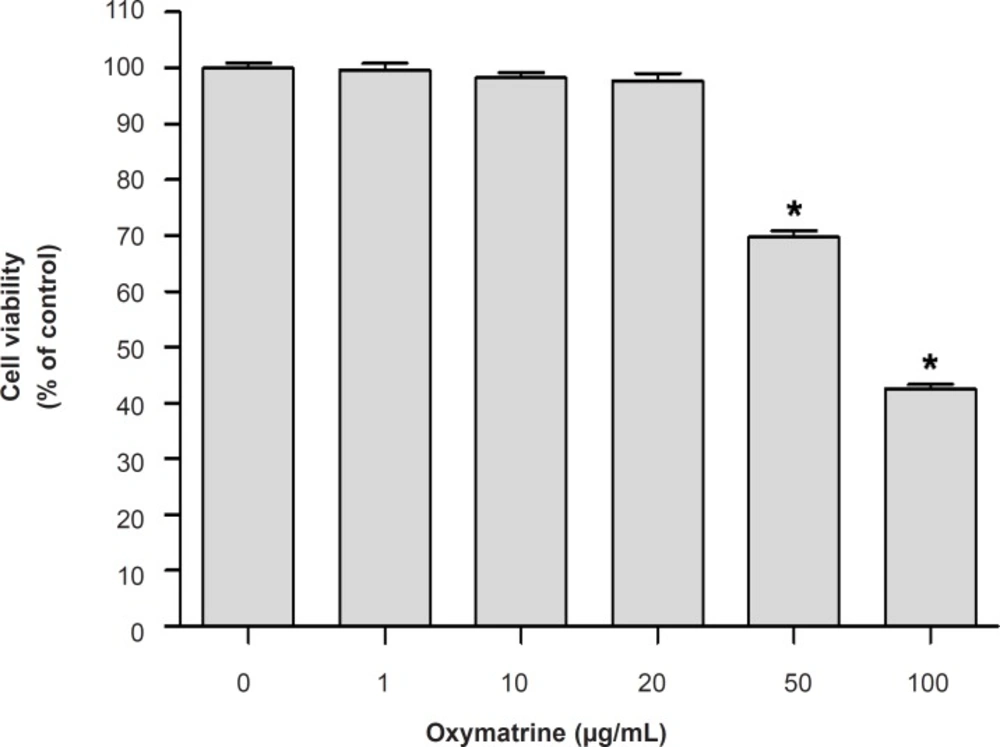
The cell viability of the cultured cells was determined by measuring the reduction of 3-(4, 5-dimethylthiazol-2-yl)-2, 5-diphenyltetrazolium bromide (MTT) to formazan. Briefly, cell were seeded and treated with different concentrations of OMT (1, 10, 20, 50 and 100 μg/mL) and 0.5 mg/mL amount of MTT solution was added to each well. After incubation for 2 h at 37°C and 5% CO2, the supernatants were removed and the formed formazan crystals in the viable cells were dissolved in dimethyl sulfoxide. The absorbance at 570 nm was determined using a microplate reader (Molecular device, USA). Data were expressed as percent change of controls. Each experiment was performed in three independent experiments.
Statistical analysis
All data in this study were presented as mean ± standard error. Statistical analysis was performed with SPSS 10.0 software (SPSS Inc., Chicago, USA). All data were analyzed by one-way analysis of variance followed by Least-significant Difference test. Significance levels were set at P<0.05.