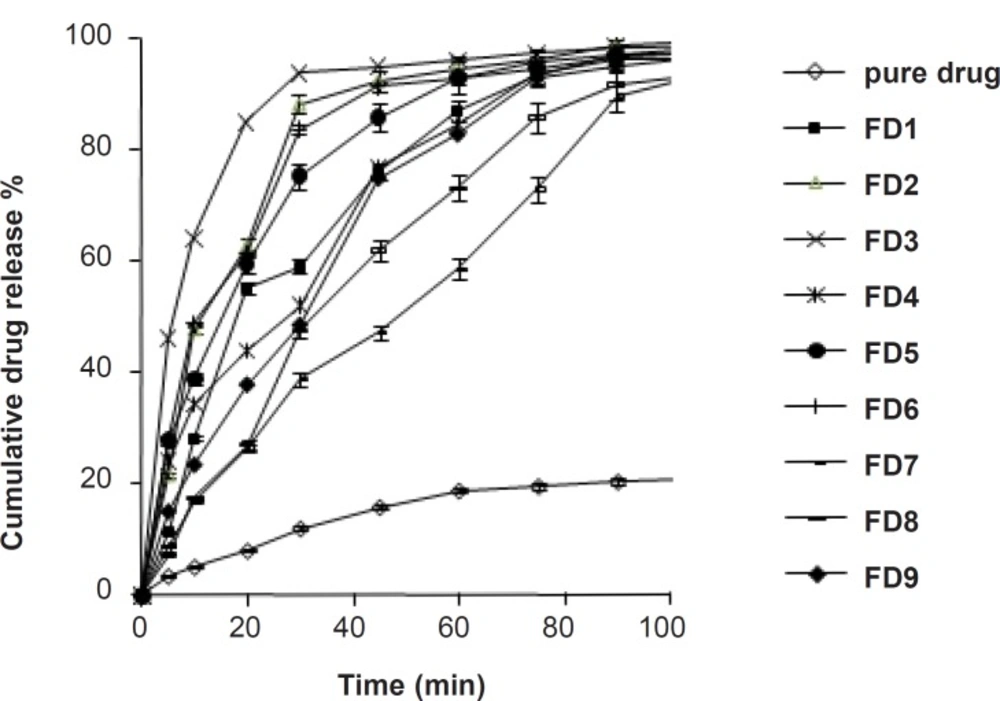
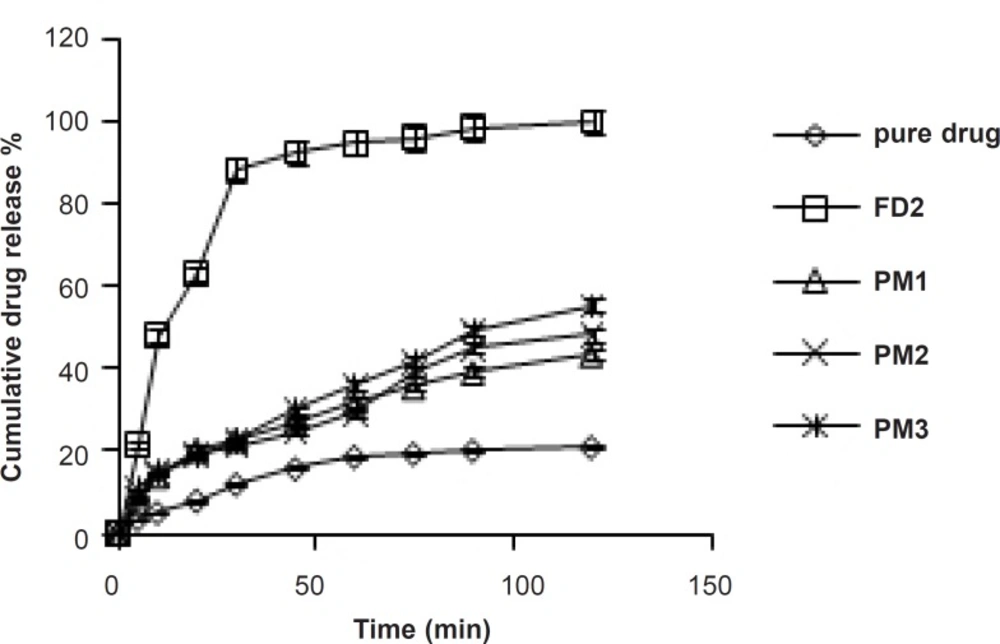
In-vitro dissolution studies
The dissolution profile of pure simvastatin was extremely low, with only 21.04% of drug release during 120 min of dissolution run in phosphate buffer (pH = 6.8), which might be attributed to the floating of the drug on the surface of dissolution medium (
15). Fused dispersions of simvastatin showed enhancement of drug dissolution due to the conversion of simvastatin into a less crystalline and/or amorphous form. The improved dissolution rate was observed in all the prepared system and maximum release was seen in FD
2 batch (99.63% in 120 min) prepared by the fusion method as shown in
Figure 1. PMs presented slight improvement in drug release which could be attributed to the improved wettability of drug particles by the presence of hydrophilic amorphous carrier as shown in
Figure 2. The model dependent parameter t
90% obtained from the cumulative percentage drug released is shown in
Table 1. The t
90% for all the nine batches (FD
1 to FD
9) displayed a wide variation from 90.65 to 21.17 min. The data clearly indicates that X
1 and X
2 strongly influence the t
90%. From the data, it was observed that on increasing melt to cool drug temperature, t
90% was increased. As the temperature was decreased, the amount of dissolved drug was increased, which may be attributed to a higher energy state of drug particles at low temperature, resulting in a more amorphous form. The superior solubility of drug at low temperatures can further be attributed to the inverse thermosensitivity of Lutrol NF 127 prill surfactant which is soluble at low temperature (but gels are soluble at higher temperature). The increased dissolution rate and bioavailability for fused dispersions (FDs) of simvastatin (SIM) using PEO-PPO block copolymer may also be attributed to the increased oxyethylene content which played a major role in the solubility enhancement. This concept is further discussed in XRD studies.
Validation of experimental design
Preliminary investigations of the process parameters revealed that X1 and X2 factors highly influenced the rate of in-vitro dissolution and hence, were used for the further systematic studies. As stated in the previous section, X1 and X2 strongly influence the t90%. A polynomial equation was constructed that it would relate the effect of individual factor and the interactions between the factors through coefficients in the polynomial equation generally calculated for a response (in this case, t90%).
Y = 44.71 + 18.45 X1 – 18.87X2 + 1.76X1X2 + 12.77 X12 + 17.31X22
The transformed Equation was:
Y = 44.71 + 18.45X1 – 18.87X2 + 12.77 X12 + 17.31X22
The fitted polynomial equations (full and reduced model) relating the response t
90% to the transformed factors are shown in
Table 2. The polynomial equations can be used to draw the conclusions after considering the magnitude of coefficient and the mathematical sign it carries (positive or negative). The significance level of coefficient b
12 was found to be p = 0.3765 and hence it was omitted from the full model equation to generate the reduced model equation.
Table 2 shows the results of regression analysis. The coefficients b
1, b
2, b
11, and b
22 were found to be significant at p < 0.05 and thus, were retained in the reduced model. Multiple linear regression analysis (
Table 2) revealed that coefficient b
1 is positive and b
2 is negative. This indicates that on increasing X
1, t
90% is increased. It was observed that by decreasing the temperature, the amount of dissolved drug is increased, which may be attributed to a higher energy state of drug particles at low temperature, resulting in a more amorphous form. The released studies of batches with increasing concentration of Lutrol NF 127 prill surfactant (X
2) revealed that as the concentration of Lutrol NF 127 prill surfactant is increased, t
90% is decreased (
Table 2). This could be due to the fact that simvastatin may exist in the fused dispersion in two different forms, namely crystalline and amorphous. The rate of drug dissolution from the fused dispersion depends on the proportion of two forms, which inturn depends on the proportion of Lutrol NF 127 prill in the fused dispersion. As the weight fraction of Lutrol NF 127 prill is increased, the proportion of the amorphous form of simvastatin may be increased, which inturn results in the enhancement of simvastatin dissolution (
16). Check-point batches FD
10 and FD
11 were prepared at X
1 = - 0.5 and + 0.5 and X
2 = + 0.5 and - 0.5 levels, respectively (
Table 1). Yates algorithm was used for the calculation of predicted response (Y
90%). The theoretical t
90% of batches FD
10 and FD
11 were 26.39 and 58.46 min, respectively. The experimental values are 28.31 and 57.67 min (
Table 1), which are in good agreement with the theoretical values. Formulations FD
2 and FD
3 that exhibited the least t
90% values, were analyzed for the selection of optimized formulation. The t
90% of both these batches were almost similar (
Table 1) and exhibited an insignificant difference as confirmed by Student t-test (tcal = 0.979, ttab = 2.78). However, the %yield of FD
2 was higher as compared to FD
3. This guided the selection of FD
2 as the optimized batch for the dissolution enhancement of simvastatin which was further subjected to the spectral characterization and pharmacodynamic activity.
Comparative in-vitro dissolution profile of pure drug and different formulations in phosphate buffer (pH = 6.8)
FT-IR studies
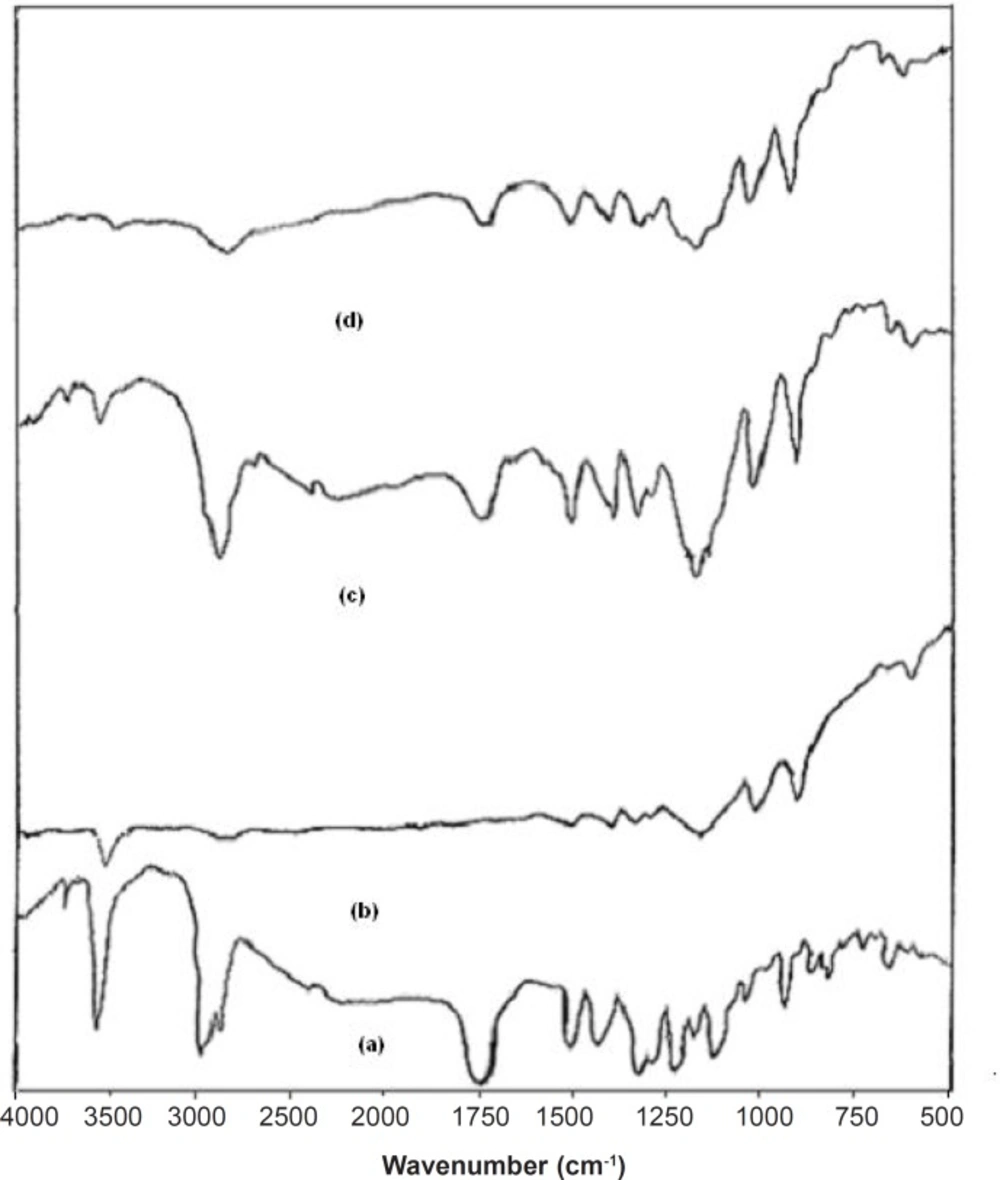
FT-IR studies were done to detect the possible interactions between the SIM and carrier (Lutrol NF 127 prill surfactant) in the fused dispersions leading to the amorphous state of simvastatin shown in
Figure 3. The FT-IR spectra of pure simvastatin presented characteristic peak at 3552 cm
-1 and 3749 cm
-1 (O−H stretch vibration), 2960 cm
-1 (C−H stretch vibration) and 1730 cm
-1, and 1164 cm
-1 and 1066 cm
-1 (stretch vibration of −C−O and −C=O carbonyl functional group). The spectrum of Lutrol NF 127 prill surfactant showed important bands at 3634 cm
-1 (O−H stretch vibration) that was attributed to the presence of water confirming the broad endotherm detected in DSC experiments and 1282 cm
-1 (−C−O stretch vibration). The FT-IR spectra of physical mixture (PM) seemed to be only a summation of drug and carrier. This result suggested that there were no interactions between the drug and carrier in PM and simvastatin maintained its crystallanity as observed in thermal analysis. If the drug and carrier interact, then the functional groups in the FT-IR spectra will show band shifts and broadening compared to the spectra of drug and carrier. Initial characterization of FDs by FT-IR studies indicated band shifts and broadening compared to the spectra of drug and carrier. In the FD
2 formulation, band shifts observed at 1714 cm
-1, 3446 cm
-1 and 3629 cm
-1 and broadening at 1165 cm
-1, suggested intermolecular hydrogen bonding via the −C=O group of simvastatin and O−H group of Lutrol NF 127 prill surfactant. Thus, a combination of interaction and decreased mobility of simvastatin during the preparation of fused dispersions may be the cause of stable amorphous form of drug inside the carrier (
17).
Comparative in-vitro dissolution profile of pure drug, FD2 and physical mixtures in phosphate buffer (pH = 6.8).
DSC studies
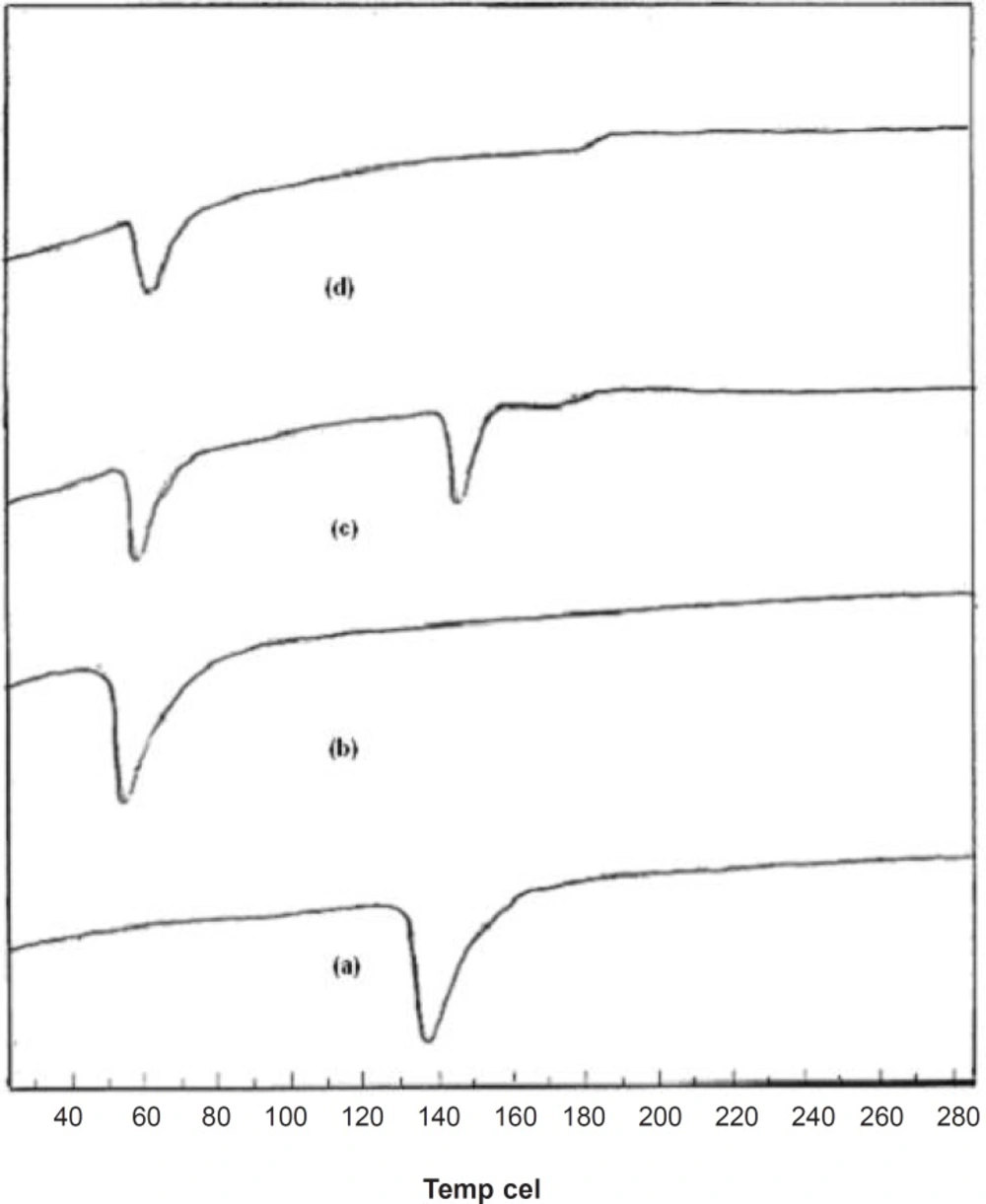
Supporting evidence for the fused dispersions was also obtained from DSC studies. DSC thermograms elicited significant suppression of carrier endothermic peak in FD
2, suggesting a homogeneous dissolution of the drug in carrier. From the thermogram, it was seen that a sharp endothermic peak corresponding to the melting point of crystalline pure drug was found at 139°C and for pure carrier at 58°C. The thermogram of the PM was merely a combination of thermogram of pure drug and carrier as shown in
Figure 4. The endothermic peak corresponding to melting of pure dug was absent in the DSC thermogram of fused dispersion of FD
2. It might be due to the presence of the amorphous form of pure drug in the fused dispersion or the dissolution of crystalline simvastatin into the molten carrier (
18). DSC thermograms of pure compounds, PM and FD
2 are shown in
Figure 4.
FT-IR spectra of (a) Simvastatin, (b) Lutrol NF 127 prill surfactant, (c) Physical mixture, and (d) FD2
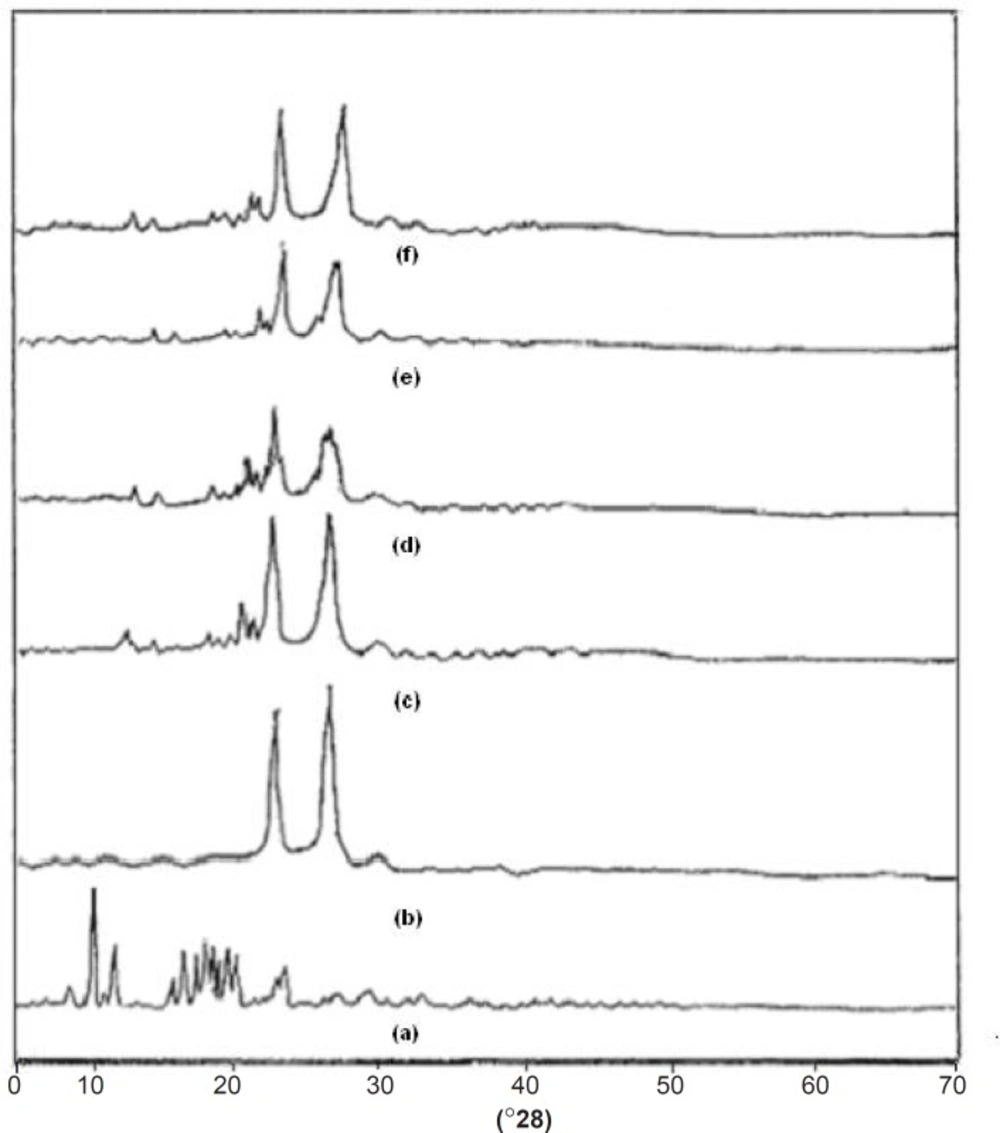
XRD studies
One of the most useful parameters for the characterization of a crystalline polymer is its degree of crystallanity (CI). Various methods have been developed for the determination of the crystallanity of polymers, one of which is x-ray diffraction analysis. The XRD pattern of pure drug showed numerous distinctive peaks in the region of 8 to 250 (2θ) (9.32, 10.88, 15.57, 16.51, 17.17, 18.75, 19.32, 22.49) that indicated the crystalline nature of simvastatin. Fused dispersions displayed all the peaks shown via the drug however, intensity of the peaks was markedly reduced with decreased d-spacing. It was observed that as the melt to cool temperature is increased, the shielding of peaks is decreased (FD
2 > FD
5 > FD
8) whereas the intensity of peaks is increased (FD
8 < FD
5 < FD
2). In case of pure carrier, the predominant peaks were observed at 19.08
0 and 23.21
0. The distinctive diffraction peaks of simvastatin in the physical mixture persisted as shown in
Figure 5. FD
2 exhibited more considerable diminution in diffraction peaks than the physical mixture. CI was found to be 71.57%, 59% and 66% for pure drug, pure carrier and physical mixture. The results also indicated that as melt to cool temperature is decreased the percentage crystallanity index decreased the FD2 (55.03%), FD5 (60.76%), and FD8 (63.71%). The significant decrease in the intensity of major SIM crystalline peaks may be due to the partial loss of crystallanity as compared with diffractograms of their corresponding PM, pure drug and FD5 and FD8 formulations. This suggested that the drug in fused dispersions is amorphous as compared to the pure drug. Hence, increased dissolution of drug was observed since an amorphous form dissolves at a faster rate owing to its higher internal energy and thermodynamic properties relative to crystalline materials (
19). X-ray diffractograms of pure compounds, PM and FD
2, FD
5 and FD
8 are shown in
Figure 5.
DSC thermograms of (a) Simvastatin, (b) Lutrol NF 127 prill surfactant and (c) Physical mixture, (d) FD2
XRD spectra of (a) Simvastatin, (b) Lutrol NF 127 prill surfactant, (c) Physical mixture, (d) FD2 (e) FD5 and (f) FD8
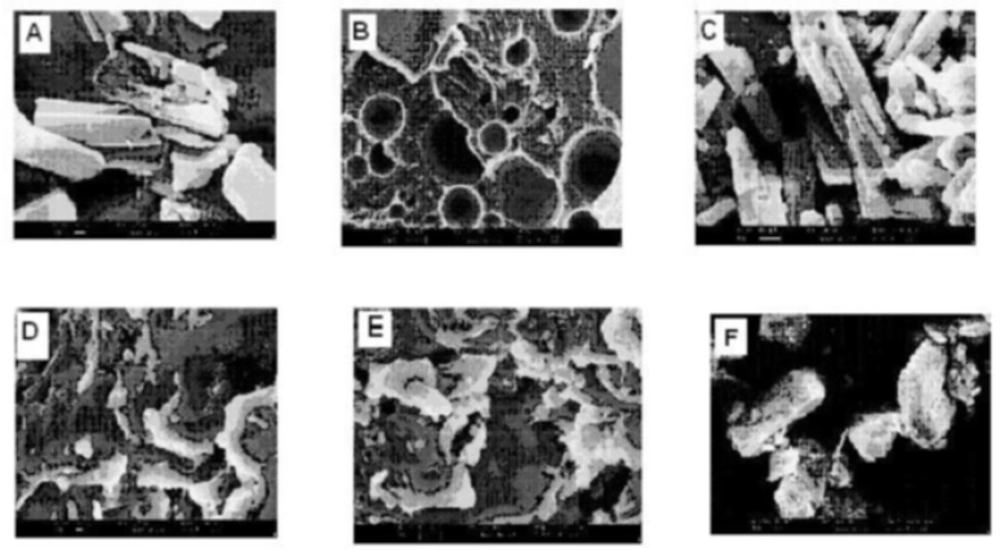
(A) SEM photomicrographs of Simvastatin, (B) Lutrol NF 127 prill surfactant, (C) Physical mixture, (D) FD2, (E) FD5 and (F) FD8
SEM studies
From scanning the electron photomicrographs, it was evidenced that the pure drug consisted of a mixture of some large crystals (8 to 10 mm) with microparticles, which might have been generated due to the micronization or any other size reduction process at the time of manufacturing. PM is seen as the combined characteristics of drug and Lutrol NF127 prill surfactant unlike the FDs where drug crystals were not possible to distinguish from carrier. FDs was formed at different melt to cool drug temperature revealed significant changes in particle shape and surface topography due to the impact of fusion process. SEM studies suggested that the homogeneous dispersions of drug in carrier may be due to the presence of amorphous state in FD
2 formulation. FD
5 and FD
8 appeared as irregular shaped agglomerates with presence of few microcrystals, suggesting the possibility of residual crystallanity. FD
5 and FD
8 appeared as the irregular shaped agglomerates with presence of few microcrystals, suggesting the possibility of residual crystallanity. Slight surface smoothness was observed in FD
5, as compared with FD
8, which could be attributed to melt to cool drug temperature difference between them. FD
2, on the other hand, looked like a smooth surface with very small particle size, suggesting the presence of amorphous state corroborating XRD observation (
20). An XRD and SEM study confirms that as the melt to cool drug temperature is decreased, the amorphization of drug is increased. SEM images of pure compounds, PM and FD
2 and also FD
5 and FD
8 are shown in
Figure 6.
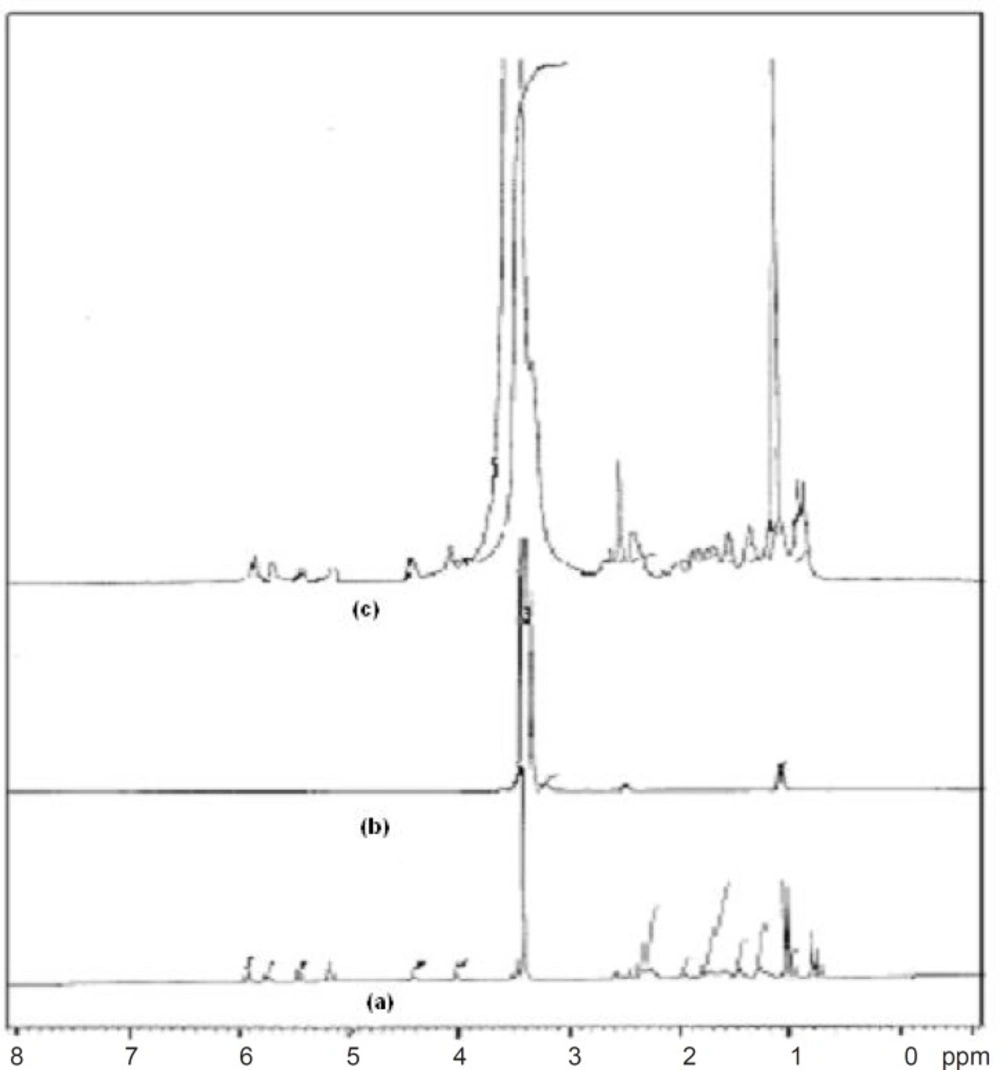
1HNMR spectra of (a) Simvastatin, (b) Lutrol NF 127 prill surfactant and (c) FD2
1HNMR spectroscopy
1HNMR spectroscopy confirmed the results obtained from FT-IR studies. The
1HNMR spectrum of pure drug showed chemical shift from 5.930 to 5.962 ppm (d,
1H, C5H), 5.736 to 5.787 ppm (t,
1H, C4H), 5.481 ppm (s,
1H, C6H), 2.257 to 2.385 ppm (m,
1H, OH), 3.456 ppm (s, 10H, C3H, C7, C2, C3, C3”, C8, C9, C10, C2”, (CH3)2, 1.495 to 1.813 ppm (m, 6H, C3”, C7”), 0.991 to 1.037 ppm (m, 6H, C4”, C5’, C4’), 1.423 to 1.470 ppm (m, 2H, C5’), 3.304 to 3.710 ppm (m, 9H, CH3) and 2.494 ppm (s,
1H, OH). Pure carrier showed chemical shift from 3.493 to 3.710 ppm (m, 89H, CH2), 1.008 ppm (s, 4H, CH3) and 3.304 to 3.439 ppm (s, 2H, CH). FT-NMR (
1HNMR) spectrum of formulation FD2 showed similar peaks of drug and carrier, 5.937 ppm (d,
1H, C5H), 5.589 to 5.767 ppm (t,
1H, C4H), 5.447 ppm (s,
1H, C6H), 2.301 to 2.388 ppm (m,
1H, OH), 3.462 to 3.729 ppm (m, 89H, CH
2), 3.462 ppm (s, 10H, C3H, C7, C2, C3, C3”, C8, C9, C10, C2”, (CH
3)
2, 1.523 to 1.845 ppm (m, 6H, C3”, C7”), 0.980 to 1.022 ppm (m, 6H, C4”, C5’, C4’), 1.022 ppm (s, 4H, CH
3) 1.445 to 1.489 ppm (m, 2H, C5’), 3.301 to 3.594 ppm (m, 9H, CH
3) and 3.336 ppm (s, 2H, CH) which confirms the intermolecular hydrogen bonding between the drug and carrier (
21).
1HNMR spectra are given in
Figure 7.
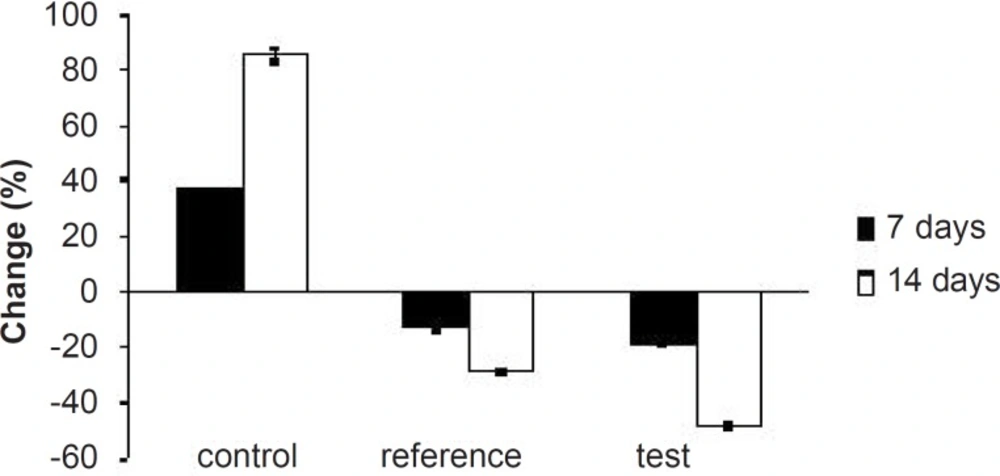
Percentage of changes in serum total cholesterol levels of experimental groups at different time intervals
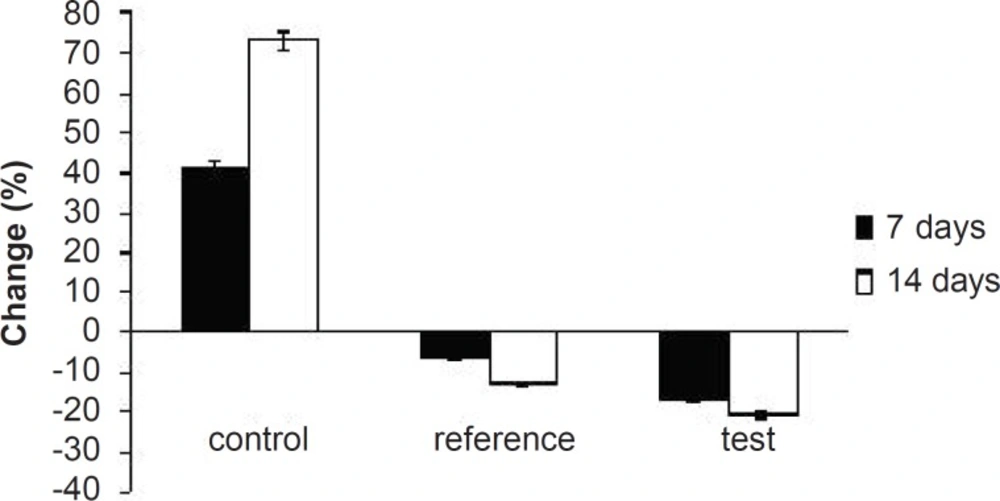
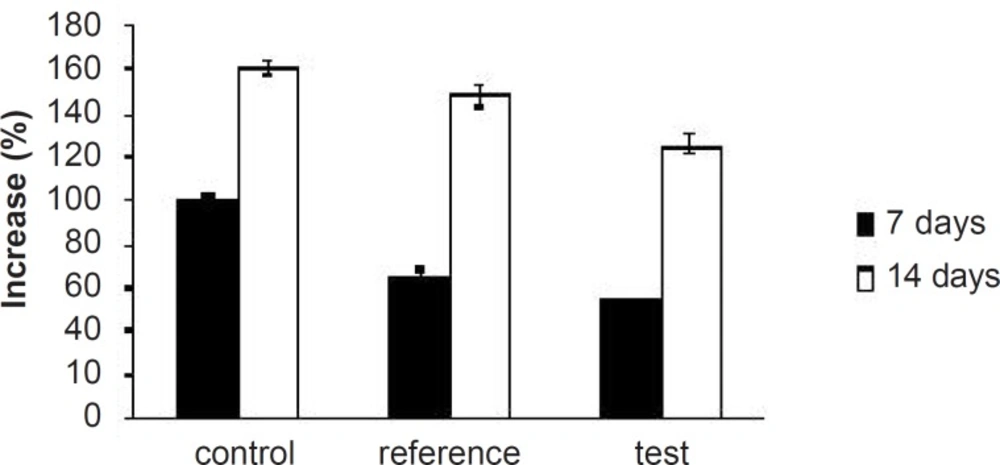
Percentage of increase in serum TG levels of experimental groups at different time intervals
Pharmacodynamic evaluation
Hyperlipidemia is an elevation of one or more of the plasma lipids, including cholesterol, cholesterol esters, triglycerides and phospholipids, in which statins play an important role for the treatment. Hyperlipidemic drugs like SIM (HMG-CoA reductase inhibitor) are known to reduce the elevated total cholesterol (TC), triglycerides (TG), LDL-cholesterol levels and VLDL-cholesterol levels in hyperlipidemic conditions. At the same time, they cause the elevation of HDL-cholesterol levels, which promote the removal of cholesterol from peripheral cells and facilitate its delivery back to the liver (
22). This pharmacodynamic effect is reported to be dose-dependent hence, used as a basis for the comparison of
in-vivo performance of pure SIM and FD
2. The administration of excess coconut oil, which is a rich source of saturated fatty acids, promotes the biosynthesis of cholesterol in liver and leads to hypercholesterolemia. The serum lipid profiles of all the experimental groups at different time intervals are presented in
Table 3. As expected, after 7 days of treatment with excess coconut oil, control group showed significant increase in total cholesterol, TG, LDL, VLDL and HDL cholesterol; whereas, the reference group showed around 7% decrease in total cholesterol, 66% increase in TG, 13% decrease in LDL, 9% decrease in VLDL and 94% increase in HDL cholesterol.
Percentage of changes in LDL levels of experimental groups at different time intervals
Interestingly, test group in comparison with the reference one presented 2.5-fold decrease in total cholesterol, 1.5-fold increase in TG, 1.4-fold decrease in LDL, 2.4-fold decrease in VLDL and 1.3-fold increase in HDL-cholesterol. After 14 days of similar treatment, control group displayed further increase in all the lipid levels; whereas, reference group showed further decrease in total cholesterol, LDL and VLDL, significant increase in TG and slight increase in HDL cholesterol. Test group on the other hand, presented 1.5-fold decrease in total cholesterol, negligible increase in TG, 1.4-fold decrease in LDL, 1.6-fold decrease in VLDL and 1.3-fold increase in HDL-cholesterol compared to the reference group. Thus, at the end of 14 days of the study, FD
2 was better in reducing the total cholesterol, TG and LDL levels than the pure SIM
(Figure 8-10). This could be primarily attributed to the improved solubility and dissolution associated with the amorphization of the drug (
10). Moreover, pharmacodynamic evaluation in rats also justified the improvement in therapeutic efficacy of optimized FDs over the pure SIM. This may attributed to the improved solubility and dissolution associated with the amorphization of the drug. Since the FD
2 batch elicited superior results, it can be proposed as a good candidate for systemic product development.