
Introduction
Experimental
Results
| Groups | Δ STa(during CO*) | Δ ST (1 h**) | Δ ST (2 h***) | Heart rate (1 h**) | Heart rate (2 h***) |
|---|---|---|---|---|---|
| 3000 PPM CO | 6 × 10-5 ± 2 × 10-5 | -5×10-5±2×10-5 | -6×10-5 ± 3 × 10-5 | 179 ± 41.2 | 221.8 ± 66.5 |
| 3000 PPM C(O+ EPO) | 4 × 10-5 ± 3 × 10-5 | -4 × 10-5 ± 3 × 10-5 | -3 × 10-6 ± 2 × 10-5 (NS) | 168.8 ± 45.2 | 181.2 ± 22 |
| 1000 PPM CO | 0 | -1 × 10-5 ± 3 × 10-5 (p < 0.05b) | -6 × 10-6 ± 6 × 10-6 (NS) | 194 ± 49.6 | 202 ± 59.8 |
| 1000 PPM CO +EPO | 0 | -2×10-5±1×10-5 (NS) | 0 | 178 ± 28 | 202 ± 24 |
| 250 PPM CO | 1 × 10-5 ± 4 × 10-6 (NS) | -2 × 10-5 ± 5 × 10-6 (NS) | -3×10-5±2×10-5 | 248 ± 67 | 203 ± 54 |
| 250 PPM CO+EPO | 0 | 0 | 0 | 250 ± 38.3 | 218 ± 39.9 |
| EPO | 0 | -4 × 10-6 ± 6 × 10-6 (NS) | 0 | 265 ± 26 | 238 ± 33 |
| Control | 0 | 0 | 1 × 10-5 ± 3 × 10-6 (NS) | 208 ± 25 | 200 ± 31 |
| Groups | Mean ± SD | Range (%) |
|---|---|---|
| 3000 PPM | 70 ± 8 | 60-76 |
| 1000 PPM | 31 ± 11 | 19-46 |
| 250 PPM | 10 ± 5 | 13-10 |
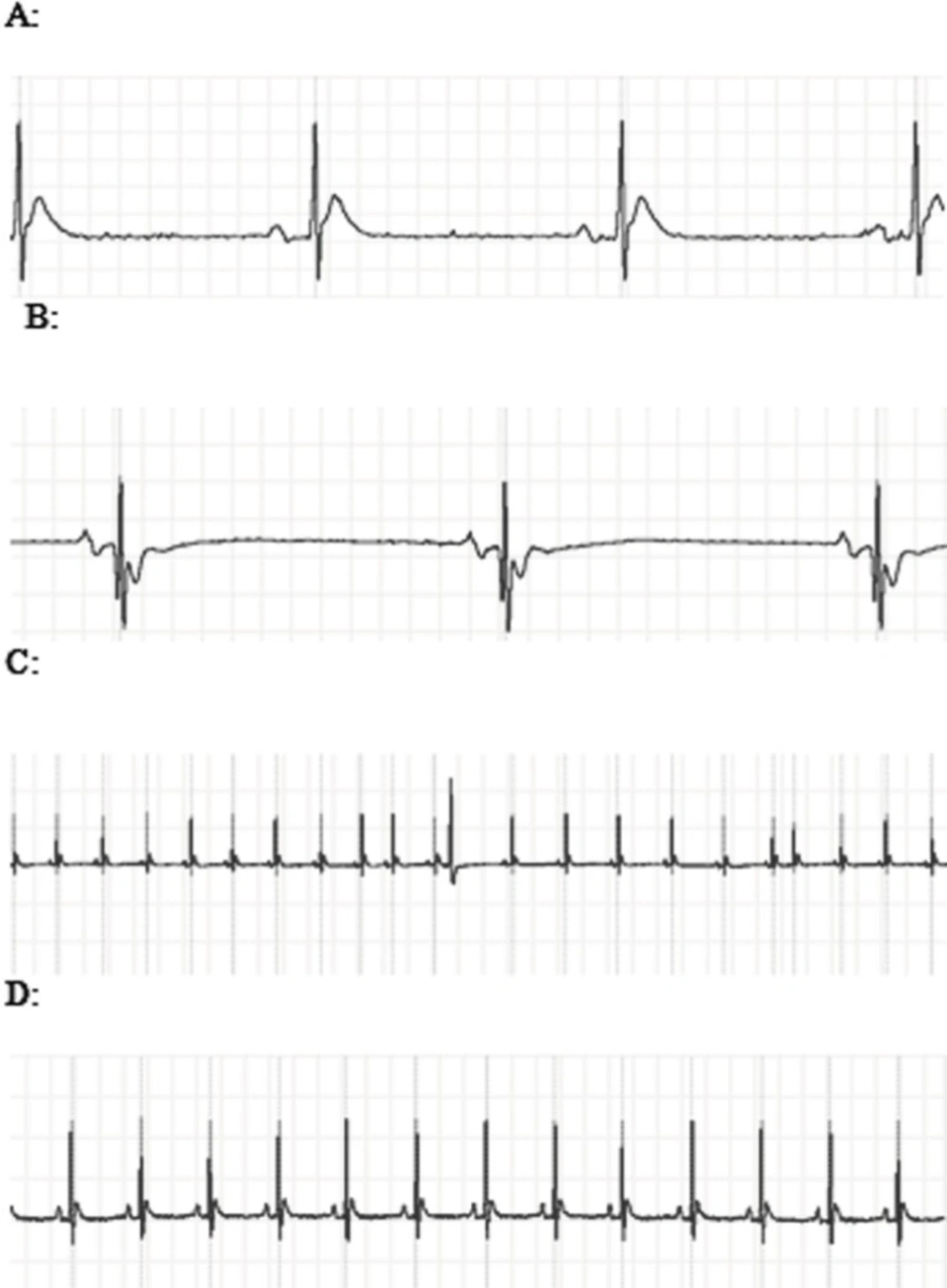
A: The ECG showed ST segment elevation during exposure to 3000 PPM of CO. B: ST segment depression and T wave inversion after 1 h of re-oxygenation following the intoxication with 3000 PPM of CO. C: PVC and sick sinus syndrome (SSS) after intoxication with 3000 PPM of CO. D: Normal
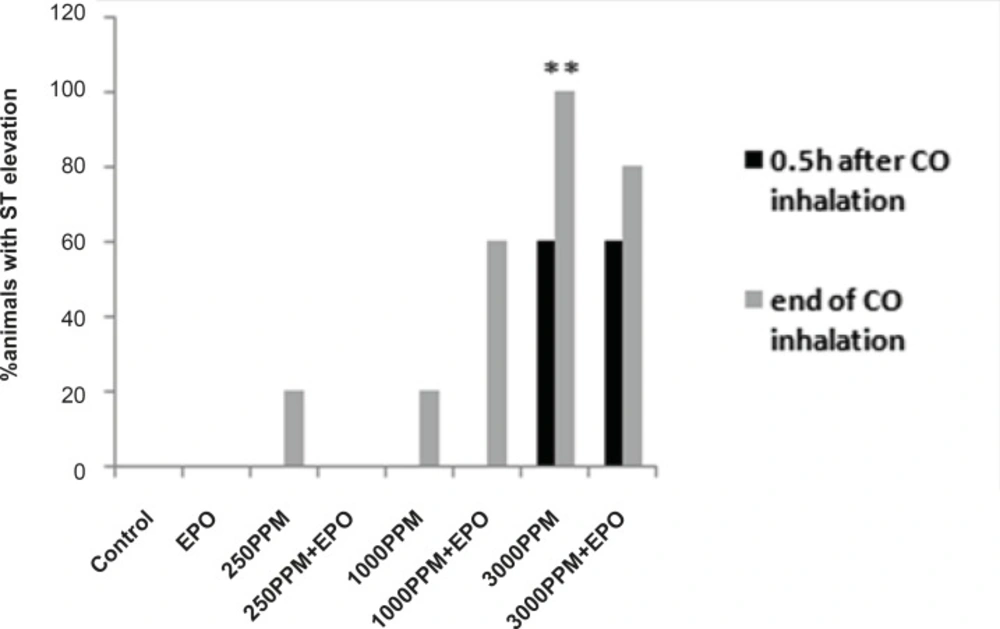
Percent of animals with ST segment elevation during CO exposure, ST elevation presented significantly in animals intoxicated with 3000 PPM of CO (**: p < 0.01 vs. control).
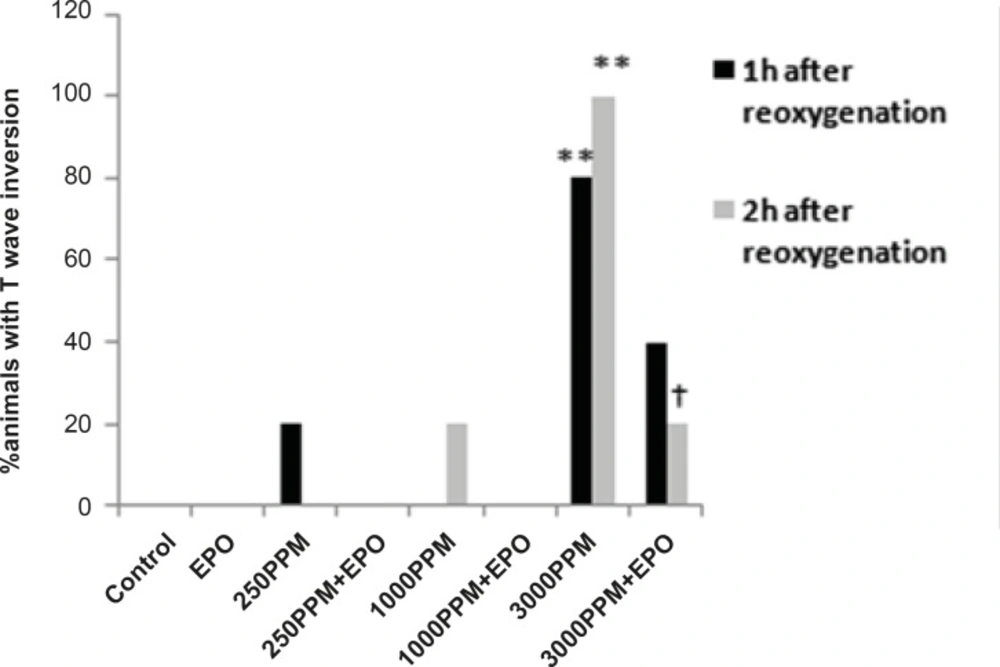
Percent of animals with T wave inversion 1 and 2 h after the re-oxygenation. This change was significant in animals intoxicated with 3000 PPM of CO after 1 h and 2 h of re-oxygenation (**: p < 0.01 vs. control). EPO significantly inhibited T wave inversion 2 h after the re-oxygenation (†: p < 0.05 vs.3000 PPM/2h after the re-oxygenation).
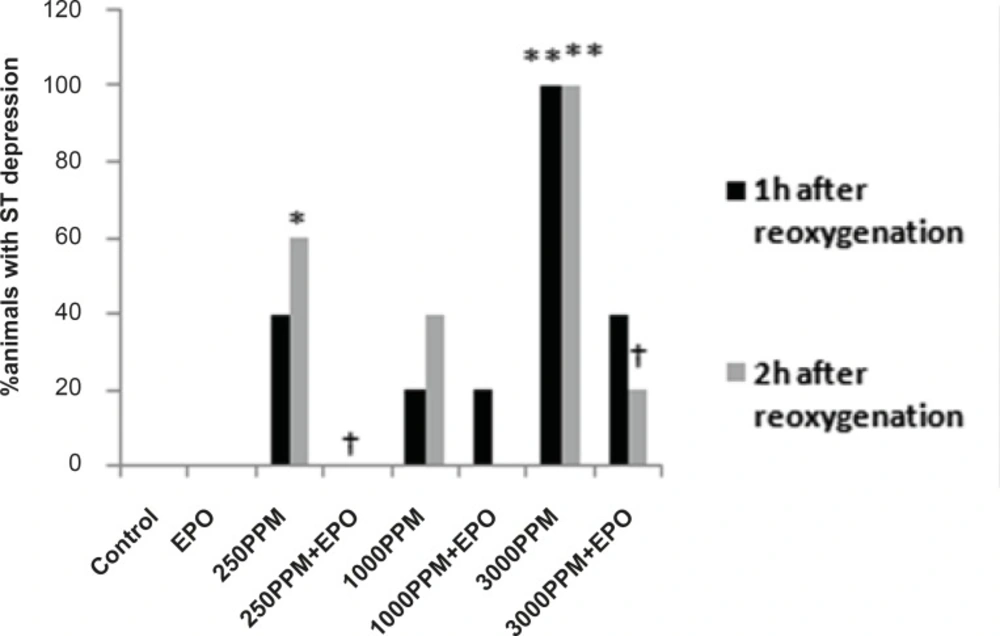
Percent of animals with ST segment depression following 1 and 2 h re-oxygenation. This change was significant in the 3000 PPM group (**: p < 0.01 vs. control) and in the 250 PPM CO group (*: p < 0.05 vs. control). EPO inhibited ST segment depression in both the 3000 (†: p< 0.05 vs. 3000 PPM) and 250 PPM groups (†: p < 0.05 vs. 250 PPM after 2 h re-oxygenation).
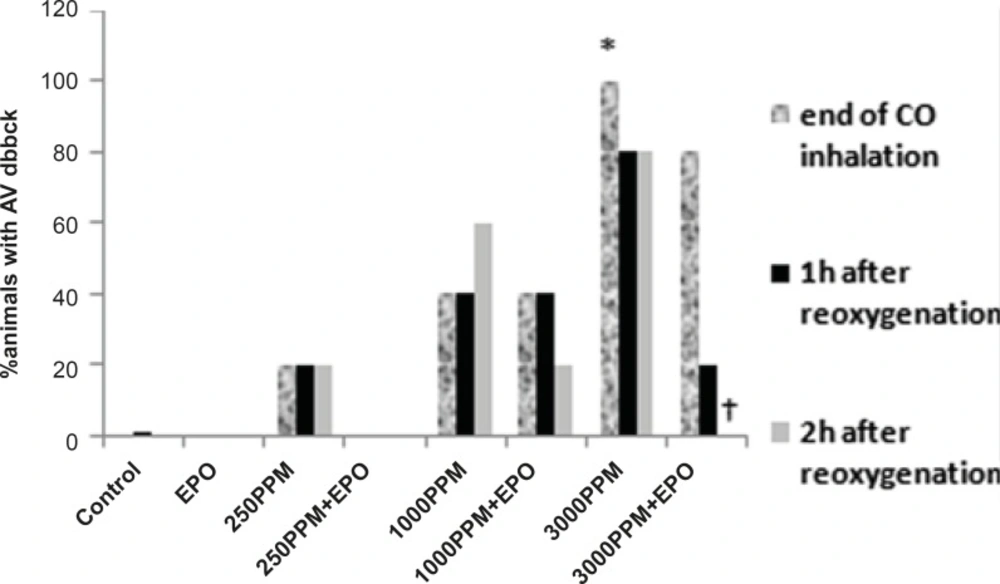
Effects of CO on PR prolongation (percent of animal with prolonged PR). PR prolongation (first degree AV block) was induced significantly following the intoxication with 3000 PPM of CO (*: p < 0.05 vs. control). First degree of AV block reduced significantly in EPO administrated animals, 2 h after the re-oxygenation (†: p < 0.05 vs. 3000 PPM 2 h after re-oxygenation).

