
Introduction
For poorly soluble, highly permeable (Class II) drugs, the rate of oral absorption is often controlled by the dissolution rate in the gastrointestinal tract (1). Therefore, together with the permeability, the solubility and dissolution behaviour of a drug are key determinants of its oral bioavailability (2). Over the years, various techniques have been employed to enhance the dissolution profile and, in turn, the absorption efficiency and bioavailability of water-insoluble salts and polymorphic forms, the formation of water-soluble molecular complexes, the drug micronization, the solid dispersion, the co-precipitation, the liophylization, the microencapsulation, and the inclusion of drug solutions or liquid drugs into soft gelatin capsules are some of the major formulation tools which have been shown to enhance the dissolution characteristics of water-insoluble drugs, however, among them, the technique of “liquisolid compacts” is one of the most promising techniques (3-8).
Formulating liquid medications into solid compacts has been the object of many studies. Jarowski (9, 10), Spireas et al. (11, 12, 13), and Javadzadeh et al. (7, 14, 15, 16, 17) worked on producing solid solutions and liquisolid based on the concept of the blending liquid medications with selected powder excipients to produce free flowing, readily compressible powders.
The liquisolid compacts are acceptably flowing and compressible powdered forms of liquid medications. The term “liquid medication” implies the oily liquid drugs and solutions or the suspensions of water insoluble solid drugs carried in suitable non-volatile solvent systems termed the liquid vehicles. Using this formulation technique, a liquid medication may be converted into a dry-looking, non-adherent, free flowing and readily compressible powder through a simple blending with selected powder excipients referred to as the carrier and coating materials (11, 12).
The anti-inflammatory agent indomethacin exhibits poor solubility. This undesirable physical property may increase the incidence of irritating side effects on the GI tract due to a prolong contact time with the mucosa (18). Numerous attempts (19-21) have been made to improve the dissolution rate of this widely used antirheumatic agent, with the purpose of obtaining a more rapid and complete absorption. This can be an ideal candidate for testing the potential of rapid-release liquisolid compacts. Nokhodchi and Javadzadeh studies (3, 15) showed that indomethacin liquisolid compacts which prepared using by Tween 80, propylene glycol and PEG 400, demonstrated a considerably higher drug dissolution rate than those of conventional capsules and directly compressed tablets.
In this study the effects of types of vehicle on the dissolution rate, and the released kinetics were investigated. These effects were analyzed statistically in the same ratios of two nonvolatile solvents with different drug solubility.
Experimental
Materials
Indomethacin was provided by Rouz Daru Co. (Iran). Microcrystalline cellulose (Avicel PH101, FMC Biopolymer, Ireland), nm-sized amorphous silicon dioxide (Mingtai chemical, Taiwan), and polyethylene glycol 200 (PEG 200), glycerin, starch, potassium hydrogen phosphate, sodium hydroxide and magnesium stearate, (Merck, Germany) were used.
The preparation of conventional tablet and liquisolid compacts
Indomethacin conventional tablets were produced by mixing the drug with avicel-silica (ratio of microcrystalline cellulose: silica was 20:1) for a period of 10 min in a cubic mixer (Erweka, Germany). The mixture then mixed with starch (10%) as disintegrating agent for 10 min, and then the Mg stearate (1%) was mixed for 5 min. The mixture was compressed on a 10 mm punch and die using a single punch tableting machine (Korsch, Germany). Sufficient compression was applied in order to produce tablets hardness of 6-7 kp, as determined through using a hardness tester (Erweka, TBH30MD, Germany). This formulation was denoted as DCT and each tablet contains 25 mg of indomethacin, 150 mg of coarse granular microcrystalline cellulose, and 7.5 mg of nm-sized silica.
Several liquisolid of indomethacin compacts (denoted as LS-1 to LS-14) were prepared as follows: Indomethacin was dispersed in PEG 200 or glycerin (these vehicles were used as the liquid vehicle to prepare the liquid medication of the different drug concentrations) with different ratios ranging from 55:45 to 60:70 (drug: PEG200/ glycerin). Then, binary mixtures of microcrystalline cellulose-silica (microcrystalline cellulose as the carrier powder and silica as the coating material with a ratio of 20 R) were added to the mixture containing the drug and PEG 200 or glycerin under continuous mixing in a mortar. Depending on the ratio of the drug and the liquid vehicle in the liquid medication used, different liquid load factor (the liquid load factor, Lf, is the weight ratio of the liquid medication and carrier powder in the liquisolid formulations.) were employed in our liquisolid preparations. These amounts of the carrier and coating materials were enough to maintain acceptable flow and compression properties. Starch (10% w/w) as disintegrant and Mg stearates as lubricant were mixed with all formulations for a period of 10 min and 5 min, respectively. The final mixture was compressed using a single punch tableting machine (Korsch, Germany). The important formulation characteristics of the prepared indomethacin liquisolid compacts are shown in Table 1.
| Liquisolid system | Liquid vehicle | Drug con. in liquid medication (% w/w) (Cd) | Liquid load factor (Lf) | Unit dose (mg) | Molecular fraction (FM) |
|---|---|---|---|---|---|
| LS-1 | PEG 200 | 55 | 0.278 | 241.2 | 0.162 |
| LS-2 | PEG 200 | 50 | 0.250 | 288.9 | 0.178 |
| LS-3 | PEG 200 | 45 | 0.227 | 346.8 | 0.198 |
| LS-4 | PEG 200 | 40 | 0.208 | 419.4 | 0.223 |
| LS-5 | PEG 200 | 35 | 0.192 | 512.5 | 0.255 |
| LS-6 | PEG 200 | 30 | 0.178 | 636.9 | 0.298 |
| LS-7 | Glycerin | 55 | 0.370 | 193.5 | 0.184 |
| LS-8 | Glycerin | 50 | 0.333 | 230.5 | 0.203 |
| LS-9 | Glycerin | 45 | 0.303 | 278.5 | 0.225 |
| LS-10 | Glycerin | 40 | 0.278 | 331.8 | 0.254 |
| LS-11 | Glycerin | 35 | 0.256 | 404.2 | 0.290 |
| LS-12 | Glycerin | 30 | 0.238 | 500.8 | 0.338 |
| DCTb | - | - | - | 202.7 | 0.000 |
Spectrophotometeric analysis
The spectrophotometeric analysis of all indomethacin samples in phosphate buffer (pH 7.2) was performed at 318 nm (UV/Visible spectrophotometer, Varian, Australia). The standard curve was constructed by serially diluting stock solution of the drug at pH 7.2 to obtain concentrations in the range of 5-60 μg/mL using phosphate buffer 7.2 as the diluent. Each sample was analyzed in three days (intraday analyzing) as triplicate.
Solubility studies
Solubility measurements were performed according to the method of Higuchi and Connors (22). In brief, solubility studies of indomethacin were carried out in buffer solution pH 7.2, PEG 200, and glycerin. Saturated solutions were prepared by adding excess drug to the vehicles and shaking on the shaker (Memmert, Germany) for 48 h at 25 ± 0.5°C under constant vibration. After this period the solutions were filtered, diluted and analyzed by UV-spectrophotometer. Three determinations were carried out for each sample to calculate the solubility of indomethacin. The solubility of indomethacin in acidic buffer (pH 1.2) was determined as well. These data showed low solubility of drug.
Dissolution studies
The USP basket method (Erweka, TD80, Germany) was used for all the in-vitro dissolution studies. The dissolution was studied in acidic buffer (pH 1.2) at first. The dissolution data showed low release (less than 6.2%), thus in this method, phosphate buffer pH 7.2 was used as a dissolution media. The rate of stirring was 100 ± 2 rpm. The amount of indomethacin was 25 mg in all formulations. The dosage forms were placed in 900 mL of phosphate buffer and maintained at 37 ± 0.1°C. At appropriate intervals (5, 10, 15, 20, and 30 min) 5 mL of the sample were taken and purified through the filter. The dissolution media was then replaced by 5 mL of fresh dissolution fluid to maintain a constant volume. The samples were then analyzed at 318 nm by UV/Visible spectrophotometer. The mean of at least four determinations was used to calculate the drug release from each of the formulations.
For assessment and comparison, drug dissolution rates (DR) were used. To this end, the amount of the dissolved drug (in μg) per minute during the first 10 min, was calculated as follows:
DR = (M × D)/1000
Where M is the total amount of indomethacin in each tablet (in this study it is 25000 μg) and D denotes percentage of the dissolved drug during the first 10 min.
Differential scanning calorimetery
Thermograms of samples (indomethacin, excipients, physical mixture of directly compressed tablets and liquisolid formulations: LS-2 and LS-8) were recorded on a DSC-60 (Perkin elmer). Samples (3-5 mg accurately weighed to 0.01 mg) were placed in aluminum pans and the lids were crimped using a Perkin elmer crimper. Thermal behaviour of the samples was investigated at a scanning rate of 10°C min-1, covering a temperature range of 30 -300°C. The instrument was calibrated with an indium standard.
Statistical analysis
All the data were statistically analyzed by analysis of variance followed by Tukey’s multiple comparison tests. A linear regression analysis was used to test associations between two parameters. Results are quoted as significant where p < 0.05.
Results and Discussion
Indomethacin was selected as the model drug for this study, since it is a slightly water-soluble substance and, thus, an ideal candidate for testing the potential of rapid-release liquisolid compacts. In addition, it can be easily assayed and quantitated in solution using spectrophotometeric principles and procedures. Beer’s law was obeyed through all the standard curves of our indomethacin solutions which were linear in the concentration range tested. The results of solubility measurements are presented in Table 2. The solubility of indomethacin (pKa = 4.5) in pH 7.2 buffer solution medium at 25°C was found to be 734 μg/mL therefore, according to the USP solubility definition, indomethacin can be considered as a slightly soluble drug at pH 7.2. The results in Table 2 show that the solubility of indomethacin is considerably increased in presence of PEG 200 and glycerin. The increase in the solubility of indomethacin at 25°C in PEG 200 and glycerin was about 121.7 and 138.3 fold respectively compared to pure indomethacin at pH 7.2.
| Solvent | Solubility (g/1000 g) |
|---|---|
| Phosphate buffer (pH 7.2) | 0.734 |
| Polyethylene glycol 200 | 89.3 |
| Glycerin | 101.5 |
Figure 1 shows the dissolution profiles of indomethacin from the liquisolid compacts (LS-6 and LS-12, ratio of drug to vehicle is 3:7) and direct compressed tablet. This Figure shows that the liquisolid compacts produce a higher dissolution rate in comparison with direct compression tablet (p < 0.000) and there was no difference between release rate in LS-6 and LS-12 after 10 and 20 min dissolution study (p = 0.302 and p = 0.089 respectively). Such enhanced drug dissolution rate may be mainly attributed to the fact that this poorly-water-soluble drug is already in solution in PEG 200 and glycerin, while at the same time, it is carried by the powder particles (microcrystalline cellulose-silica) of the liquisolid vehicle. Thus, its release is accelerated due to its markedly increased wetability and surface availability to the dissolution medium.
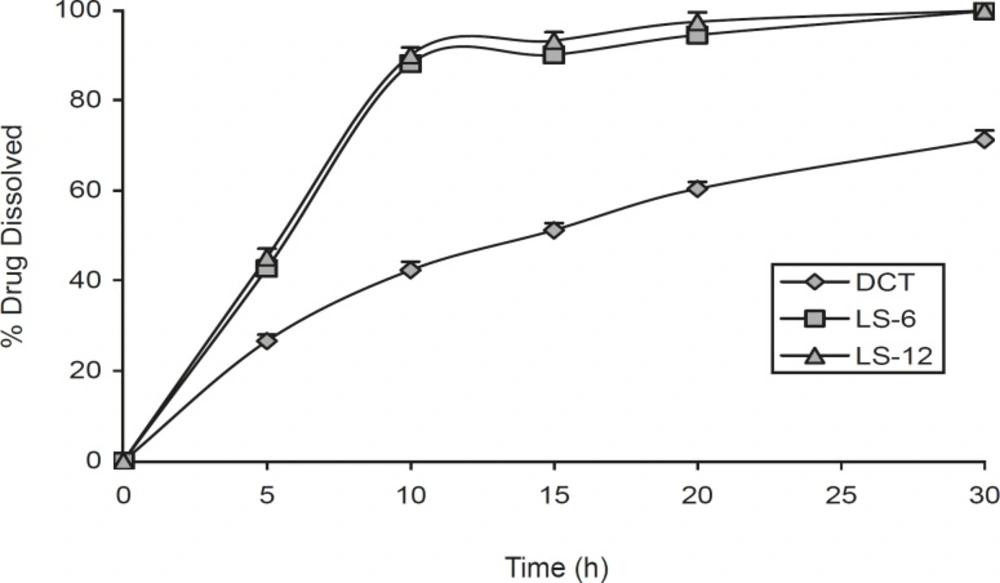
The dissolution profiles of indomethacin liquisolid compacts (LS-6 and LS-12) and directly compressed tablet (DCT). Error bars are standard deviations for at least 4 determinations
Figure 2 shows the DR of indomethacin from investigated liquisolid compacts (the highest and the lowest concentrations of PEG 200 and glycerin in formulations) and directly compressed the tablet in the first 10 min. As it is clear from this figure, the liquisolid tablets displayed better in-vitro release characteristics than those of the directly compressed tablet (p < 0.001). According to the classic dissolution equation (23):
DR = (D/h) S (CS – C)
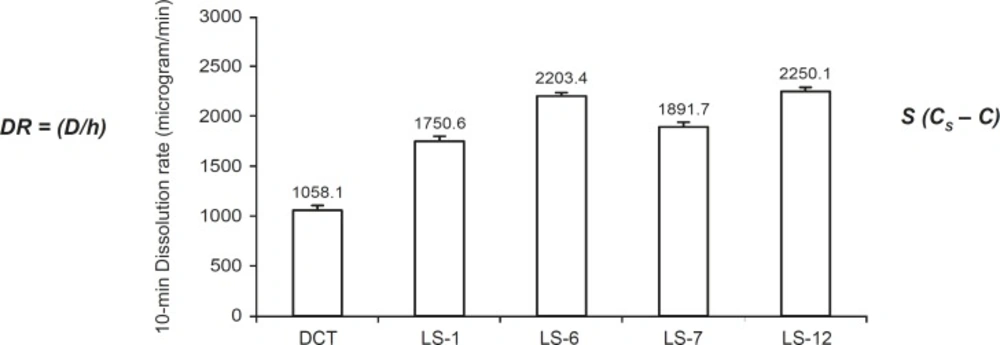
The comparison of the 10 min dissolution rate of indomethacin exhibited by liquisolid compacts containing PEG 200 (LS- 1 and LS-6) and glycerin (LS-7 and LS-12) and directly compressed tablet (DCT). Error bars are standard deviations for at least 4 determinations
The drug dissolution rate (DR) of a drug is directly proportional to its concentration gradient (CS – C) in the stagnant diffusion layer and its surface (S) available for dissolution. CS is the saturation solubility of the drug in the dissolution medium and thus, it is a constant characteristic property related to the drug and dissolving liquid involved. Since all of the dissolution tests for formulations were done at a constant rotational paddle speed (100 rpm) and identical dissolving media, we can assume that the thickness (h) of the stagnant diffusion layer and the diffusion coefficient (D) of the drug molecules remain almost identical. Therefore, the observed higher dissolution rates of indomethacin from liquisolid tablets are due to the significantly increased surface of the molecularly dispersed indomethacin (12).
Figure 3 shows the effect of the drug concentration (Cd) in the liquid medication on the 10 min dissolution rate (DR) of indomethacin from the PEG 200 and glycerin liquisolid compacts. It can be seen that, as the concentration of the drug in the liquid medication (Cd) decreased from 55% to 30% w/w, the values of DR increased. The figure shows that the drug concentration in the liquid medication is one of the main factors on the performance of a liquisolid compact and has considerable effect on the indomethacin 10 min dissolution rate. It can be seen that DR decreased with an increase in the concentration of liquid vehicle (PEG 200 or glycerin). However, when the concentration of indomethacin was increased from 50% to 55% in LS-2 and LS-1 formulations, there was no significant difference in DR values (p = 0.27). In liquisolid tablets which containing glycerin, the increase of indomethacin’s concentration from 55% to 45% in LS-7 to LS-9, showed no significant differences in 10 min DR values (p = 0.948 and p = 0.198 respectively). The same results were reported by Javadzadeh et al. (7); their study showed non significant increase in the dissolution rate, when the concentration of piroxicam was increased from 30% to 50% w/w.
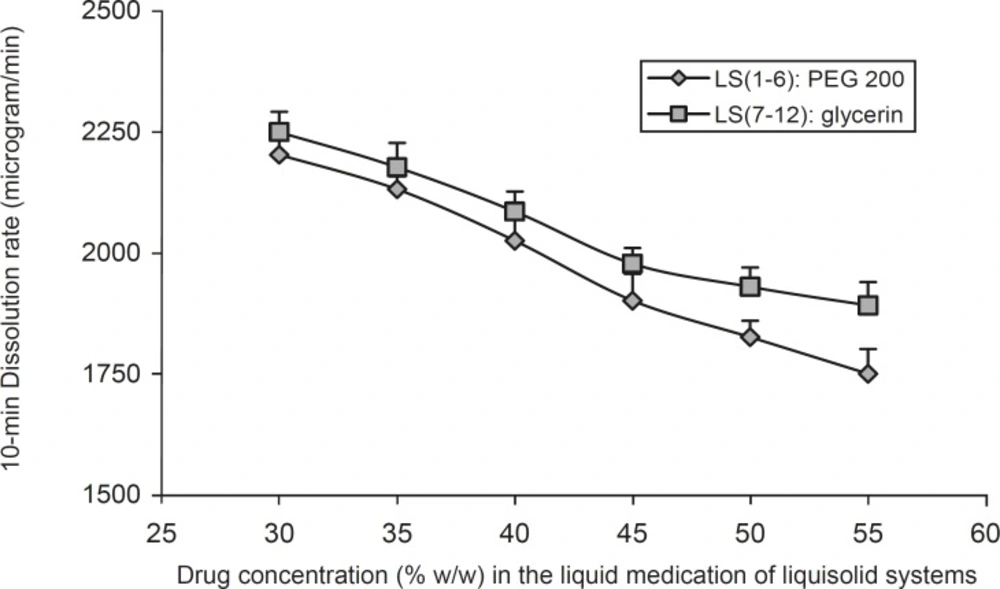
The effect of drug concentration (Cd) in the liquid medication on the 10 min dissolution rate (DR) exhibited by different liquisolid formulations. Error bars are standard deviations for at least 4 determinations
Such differences in the DR values of indomethacin from liquisolid compacts observed in Figure 3 may be justified using the differences in the amount of soluble form of the drug or molecular dispersion states of the drug in the formulations. For instance, since the saturation solubility of indomethacin in PEG 200 is 8.93% w/w (Table 2), about 40.18% of the drug is as soluble form in LS-1 formulation (this formulation contains 55% drug and 45% PEG 200); whereas in LS-6 formulation, about 62.5% of indomethacin is as soluble form (LS-6 formulation contains 30% indomethacin and 70% PEG 200). In similar formulations (LS-7 and LS-12) which contains the same concentrations of drug and the same amount of glycerin, about 45.7% and 71.1% of indomethacin is as soluble form, respectively.
In order to investigate the effect of fraction of the dissolved or molecularly dispersed of indomethacin (FM) of the prepared liquisolid tablets on the dissolution rates of the drug from liquisolid compacts and DC tablets, the FM was plotted against their corresponding DR values (Figure 4).
FM can be defined as the ratio of the drug’s saturation solubility (CL) in the liquid vehicle over the drug concentration (Cd) in the liquid medication. Therefore, FM = CL/Cd, where FM = 1 and when CL/Cd > 1. Based on the above equation, the FM values of formulations are listed in Table 1 Since no liquid vehicle is involved in the case of directly compressed tablets which contain plain indomethacin powder, its FM value was taken equal to 0.
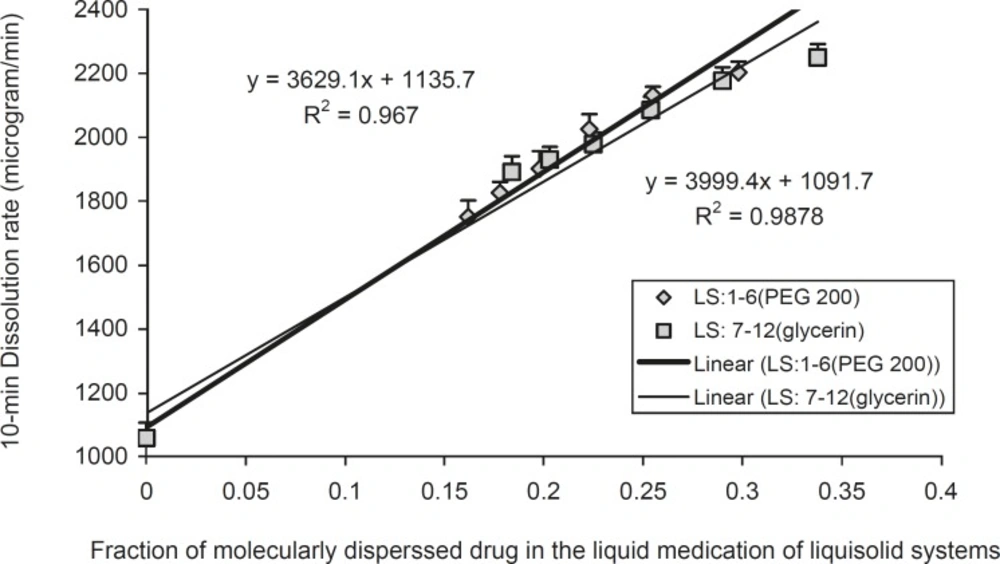
The effect of the fraction of molecularly dispersed drug (FM) in the liquisolid systems on the 10 min dissolution rate (DR) of indomethacin, exhibited by various liquisolid formulations. Error bars are standard deviations for at least 4 determinations
In Figure 4, the DR in 10 min, increased in a linear manner, with increasing FM values of the liquisolid systems can be seen. Therefore, it is possible to predict the dissolution rate (DR in μg/min) of indomethacin liquisolid compacts containing PEG 200 or glycerin liquisolid tablets, which will be obtained within the initial 10 min of the dissolution process. A plot of DR against FM (when 0.162 < FM < 0.298 for PEG 200 contains liquisolid systems and when 0.184 < FM < 0.338 for glycerin contains liquisolid formulations) shows that the DR changes linearly with FM. The high correlation coefficients of 0.993 and 0.983 provide a value that characterizes the effect of FM on indomethacin release from PEG 200 and glycerin liquisolid compacts, respectively.
As shown in Figure 4, for systems with FM values ranging 0.162-0.298 for PEG 200 containing liquisolid systems and when 0.184-0.338 for glycerin containing liquisolid formulations, the dissolution rate of indomethacin may be given by:
DR = 3999.4 FM + 1091.7
(in PEG 200 containing liquisolid systems)
DR = 3629.1 FM + 1135.7
(in glycerin containing liquisolid systems)
The effect of the type and amount of vehicle on the release rate of indomethacin from liquisolid compacts is shown in Figures 5 and 6. It can be seen from the Figures, all two vehicles used in liquisolid compacts produced a higher dissolution rate than those of conventional tablet. The evaluation of the type liquid vehicle on release rate of investigated liquisolid formulations in LS-1 and LS-2 compared to LS-6 and LS-7 (with Cd of 55% and 50% w/w), showed a higher 10 min dissolution rate in liquisolid systems containing glycerin in comparison with those containing PEG 200 with similar amounts of liquid vehicle (p = 0.007). This difference was not observed after 20 min (p = 0.054 and p = 0.189, respectively). In other formulations with similar amounts of investigated liquid vehicles, no differences between dissolution rate after 10 and 20 min were observed (p > 0.05). The relationship between the percentage release of the drug and the solubility in medium was reported previously by Nokhodchi et al. (3). The similarity of the drug solubility in PEG 200 and glycerin caused small differences in drug dissolution rate in this investigated formulations.
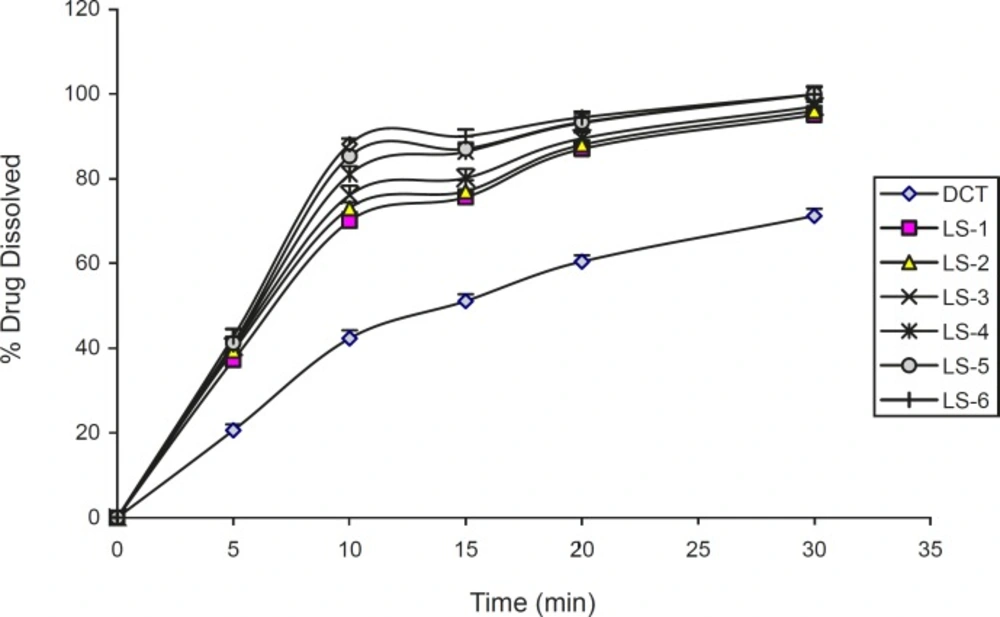
The dissolution profiles of indomethacin from liquisolid compacts containing different amounts of PEG 200. Error bars are standard deviations for at least 4 determinations
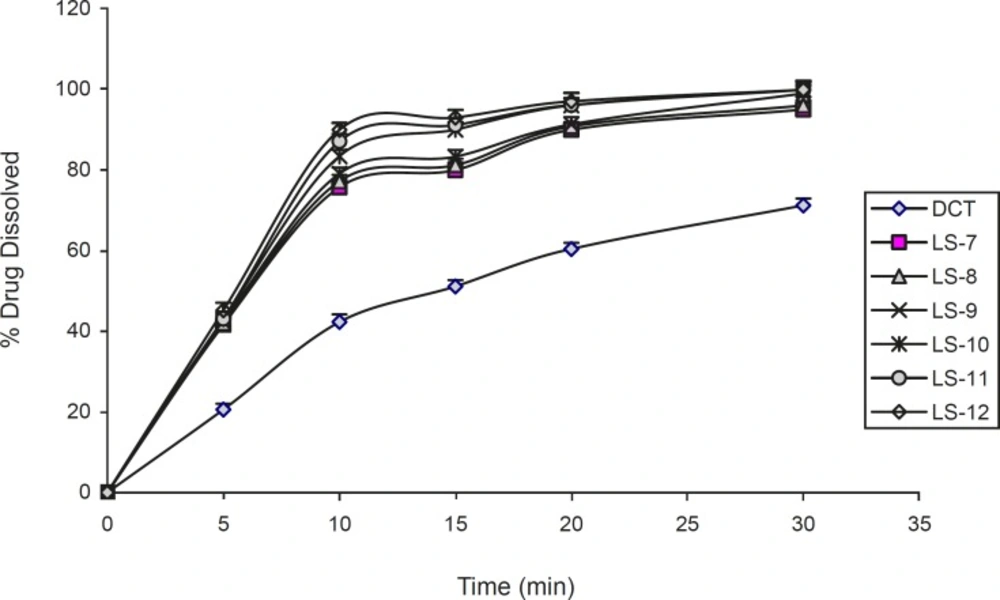
The dissolution profiles of indomethacin from liquisolid compacts containing different amounts of glycerin. Error bars are standard deviations for at least 4 determinations
DSC thermo grams are shown in Figure 7. Indomethacin showed an endothermic peak around its melting point. The liquisolid and physical mixture formulations showed the same peak in this area which indicates that there is no interaction between the drug and excipients or changes in crystallinity of the drug during the formulation process. From the above finding, it can be concluded that the enhanced dissolution rate of indomethacin liquisolid compacts is not due to the formation of a complex between the drug and excipients.
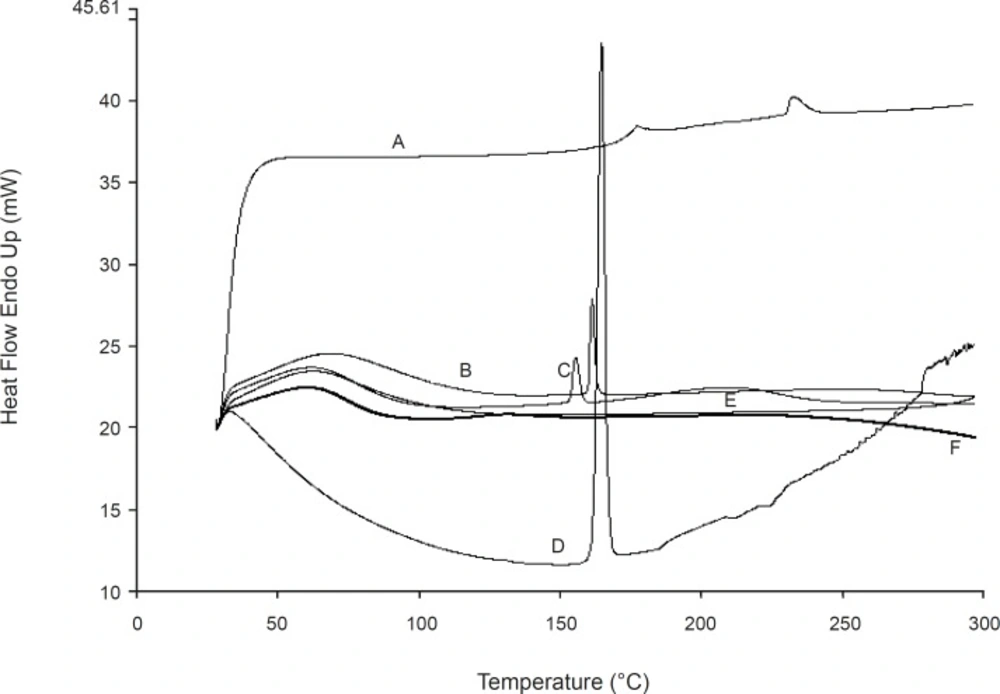
The differential scanning calorimetery of (A) silica, (B) physical mixture of directly compressed tablet, (C) LS-8 (containing glycerin), (D) indomethacin, (E) LS-2 (containing PEG 200) and (F) Avicel
Conclusion
The liquisolid compacts technique can be a promising alternative for the formulation of water-insoluble drugs, such as indomethacin into rapid release tablets. The higher dissolution rates displayed by liquisolid compacts may also imply enhanced oral bioavailability due to the increased wetting properties and the surface of drug available for dissolution. It can also be concluded that from this study, the investigated liquisolid compacts of indomethacin, in different drug concentrations in their liquid medications, exhibits the drug dissolution rates which are directly proportional to the fraction, FM, of the molecularly dispersed drug in the liquid medication.
