Ketoconazole was obtained as a gift sample from Yash Pharmaceuticals, Roorkee, India. Pluronic F127 was provided as gift sample by Signet Chemicals, India and its melting point varies from 52 to 57°C. PVP K-30 was procured from Central Drug House, New Delhi, India and gelatin capsule shells (000 sizes) were purchased from Saini Medical Hall, Punjab, India. All the materials used in the study were of analytical grades.
In order to evaluate the effect of carriers on Ketoconazole, phase solubility studies and dissolution studies were performed. Physical analysis based on DSC, FTIR, and XRD was performed to evaluate the structure of the dispersions and to detect the possible drug-carrier interactions.
Phase solubility studies
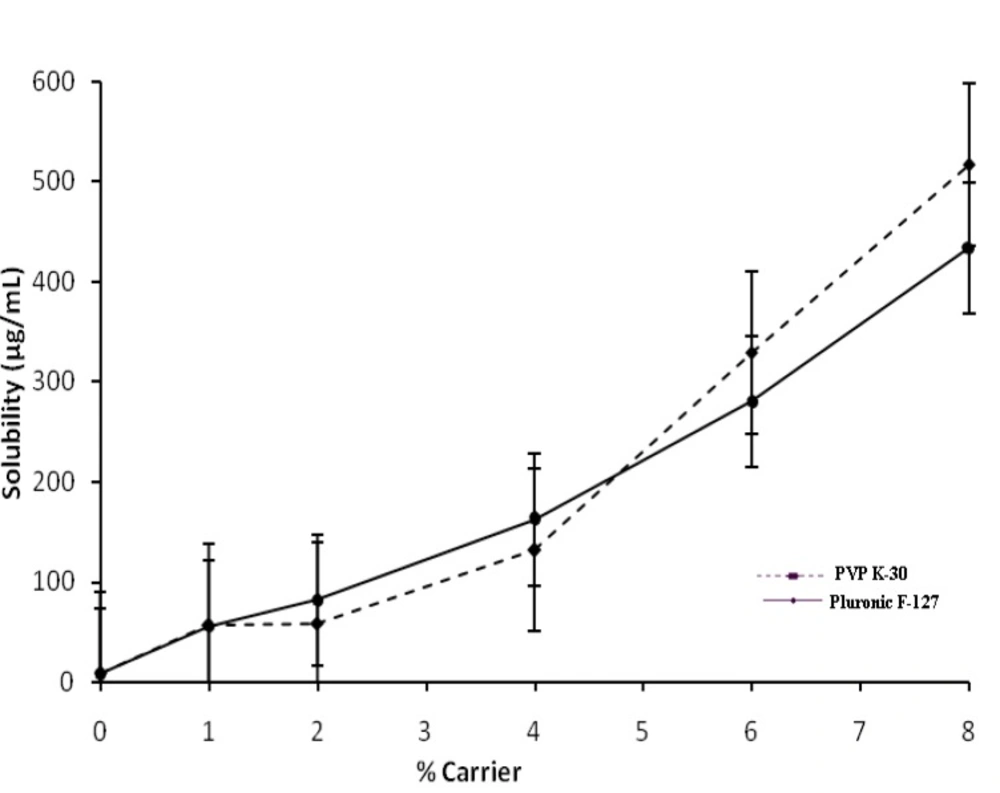
Phase solubility studies were performed (method proposed by Higuchi and Connors) (
25) by adding an excess of Ketoconazole to 50 mL volumetric flasks containing 10 mL triple distilled water, phosphate buffer pH 6.8 and aqueous carrier solutions (1%, 2%, 4%, 6% and 8% w/v concentration range) of Pluronic F127 and PVP K-30. Volumetric flasks were shaken on water bath incubator shaker (Navyug Q-5247, India) at 30 ± 0.5°C for 48 h. At equilibrium after 48 h, the suspension was filtered through whatman filter paper (0.45 μ pore size) and aliquots were withdrawn to determine drug content spectrophotometrically at λ
max244.8 nm (Systronics UV 2203 Spectro-photometer, Ahmedabad, India).
Preparation of physical mixtures
Physical mixtures (PMs) of ketoconazole were prepared by mixing drug with carrier in a mortar (
26) until homogenous mixture was obtained. The resulting mixture were sieved through a 355 μm mesh and then stored in a desiccator at room temperature until use. As Pluronic F127 is a crystalline carrier, it was first crushed and then passed through 355 μm mesh before mixing with drug.
Preparation of solid dispersions formulations
Melt-fusion method
As Pluronic F127 has a melting point range of 52-57°C (
27), solid dispersions were prepared by melt-fusion method with different concentrations of Ketoconazole (1:1, 1:2, 1:4, 1:6, and 1:8) and through melting physical mixture of drug and Pluronic F127 on a water bath at 90°C with continuous stirring and by rapidly cooling the resulting homogeneous preparations rapidly cooled over ice bath (
28,
29) Subsequently, the dispersions were pulverized, passed through a 355 μm sieve, and then stored in a desiccator at room temperature until use.
Solvent evaporation method
As PVP K-30 is soluble or sparingly soluble in a wide range of commonly used solvents like methanol, ethanol, chloroform, dichloromethane
etc. (
27), therefore, solvent evaporation method is suitable technique for preparing solid dispersions of drug with PVP K-30. Chloroform was used as a common solvent for preparing solid dispersions. Five different preparations of Ketoconazole with PVP K-30 were prepared in the drug to carrier ratios of 1:1, 1:2, 1:4, 1:6, and 1:8 respectively. Ketoconazole and carrier were dissolved in chloroform subsequently placed in a vacuum oven (Navyug Q-5247, India) at 15 lb pressure, 40°C temperature for 72 h until formulations appeared in a glassy transparent stage without any moisture or stickiness and were stored in dessicator for further 48 h. Subsequently the dispersions were pulverized, passed through a 355 μm mesh and stored in dessicator at room temperature until use.
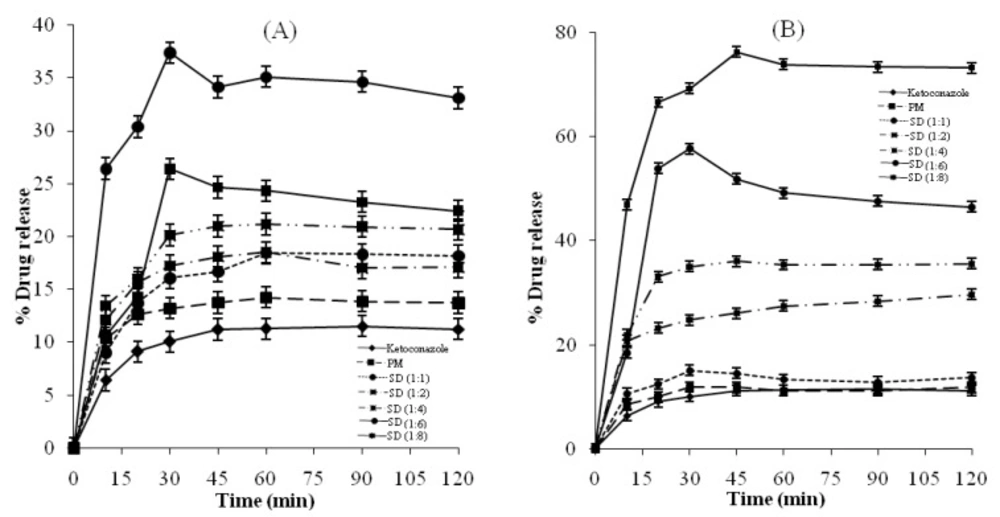
Dissolution studies
Dissolution rate studies were performed in 900 mL phosphate buffer (pH 6.8) at 37 ± 0.1°C; using single stage USP XXII apparatus (paddle method, 100 rpm). Pure Ketoconazole drug, solid dispersions as well as physical mixtures, each containing equivalent to 100 mg drug were filled in empty hard gelatin capsule shells and subjected to dissolution studies. At fixed time intervals (10, 20, 30, 45, 60, 90, 120 min) aliquots of 5 mL samples were withdrawn and simultaneously replenished with fresh 5 mL of phosphate buffer solution maintained at same temperature to maintain sink conditions. Samples, so withdrawn were immediately filtered through whatman filter paper (pore size 0.45μ) and assayed spectrophotometrically for drug content at 244.8 nm.
Content uniformity
Precisely weighed amounts of solid dispersions equivalent to 20 mg of Ketoconazole was dissolved in 50 mL methanol. The drug content was determined spectophotometrically with appropriate dilutions from methanol at 244.8 nm.
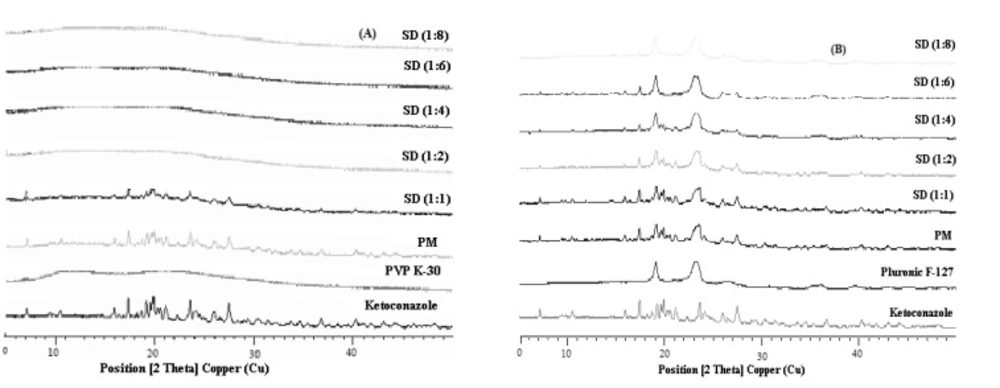
X-Ray diffraction study
Powder X-ray diffraction (XRD) can be used to qualitatively detect material with long range order. Sharper diffraction peaks indicate more crystalline material. The crystallinity of the pure drug, carriers and solid dispersions were measured using a XPERT-PRO apparatus exposed to CuKα (λ = 1.54060 Å) radiation (45 kV X 40 mA) from a PW3064 X-ray generator. The degree of diffractions was measured at 10°/min every 0.017° between 5º and 45º (2θ). Samples were prepared into aluminum frames. For the preparation, the front of the frames was mounted on a smooth teflon plate. The samples were filled into the window and were compressed with a slide. This procedure avoideda preferential orientation of the particles. Silicon was used as an internal standard.
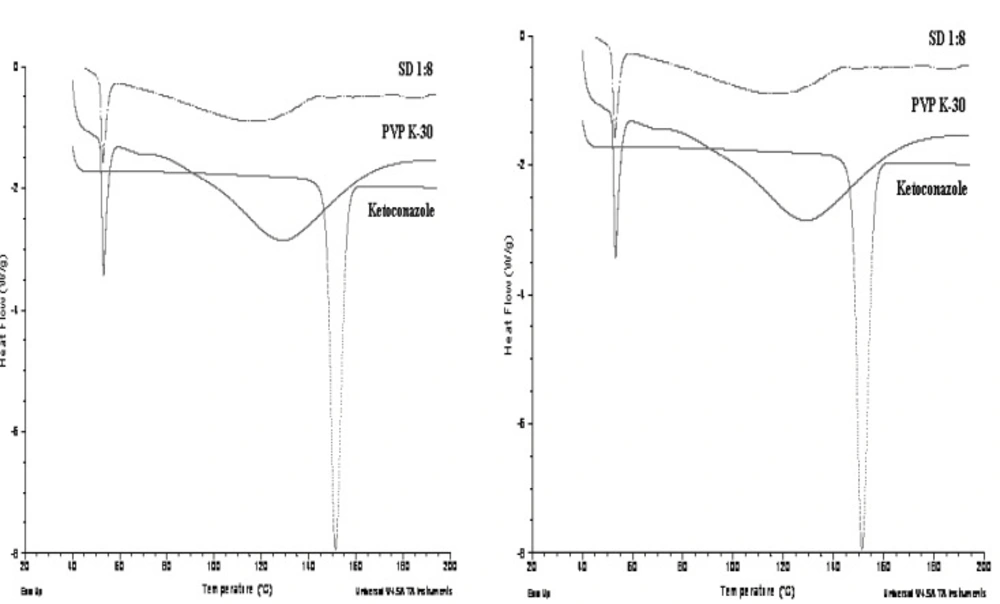
Differential scanning calorimetry study
Differential scanning calorimetry (DSC) measurements were carried out of the Ketoconazole, carriers and solid dispersion formulations which showed better release rate from dissolution studies using a DSC Q10 V9.9 (Build 303, Germany). Samples of approximately 3-4 mg were sealed in pierced aluminum pans of 40 μL and were measured at a scanning speed of 20°C /min over a temperature range from 40-200°C. The samples were cycled twice to remove the effects of moisture and thermal history. The solid-state characteristics were surveyed in the heating cycle by observing the melting enthalpy and the onset melting temperature of the drug. An empty pan served as reference.
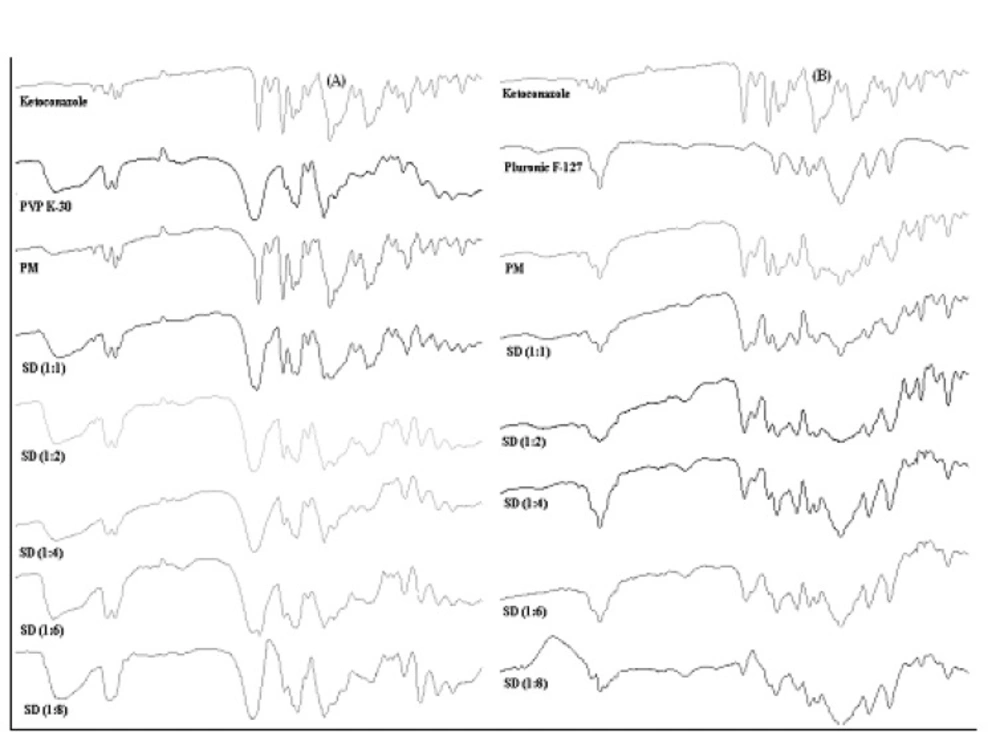
Fourier transform infrared spectroscopy
Fourier transform infrared spectroscopy (FTIR) spectra were obtained on a Shimadzu 8400S IR solution 1.30 system, (Shimadzu Corporation) using potassium bromide (KBr) disc method. To avoid the effect of moisture, all samples were dried overnight in a dessicator. The FTIR spectra, in transmittance mode, were obtained in the spectral region of 4000-400 cm-1 using a resolution of 4 cm-1 and 20 scans. A polystyrene filter was used to check the spectrophotometer calibration and KBr pellet was used as reference.