
Introduction
Phencyclidine (1-(1-phenylcyclohexyl) piperi- dine, CAS 956-90-1, PCP, I) is a semi-rigid molecule containing a cyclohexane ring with attached aromatic and piperidine rings (Figure 1). PCP and its analogues are highly potent and widely abused psychotomimetic drugs which influence the central nervous system. They display analgesic, stimulant, depressant and hallucinogenic effects due to specific binding sites in the brain (1). PCP binds to the N-methyl-D-asparate (NMDA) receptor complex and blocks NMDA-mediated gating of the calcium conductance channel (2). The analgesic effect of ketamine (2-O-chlorophenyl-2-methylaminocyclohexan, CAS 1867-66-9, II, Figure 1), another PCP analogue, was first described by Domino et al. in 1965 (3). Ketamine is a low-affinity, use-dependent, non-competitive antagonist of NMDA receptors (4-6).
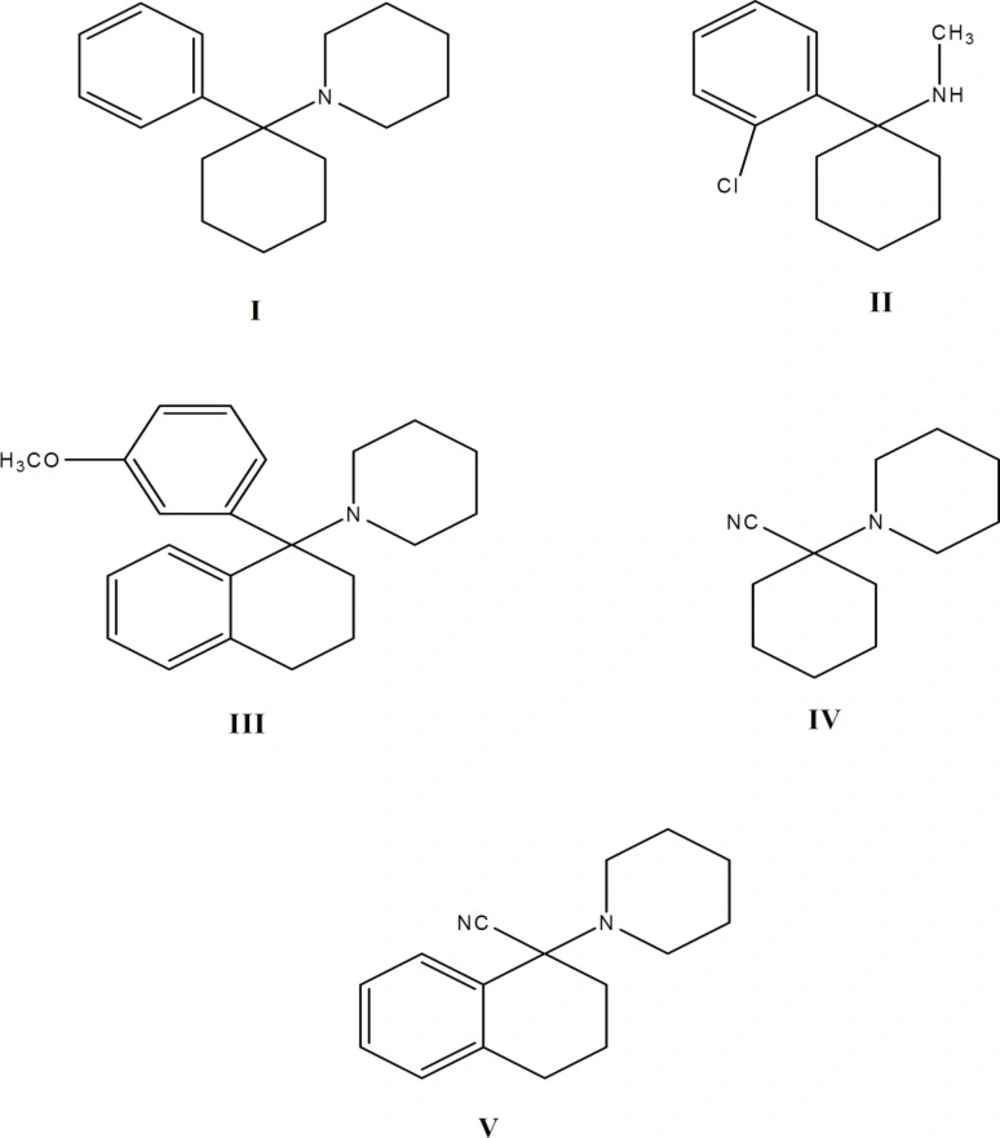
Structure formulas of PCP (I), Ketamine (II), PCP-OCH3-tetralyl (III) and Carbonitrile intermediates IV and IV
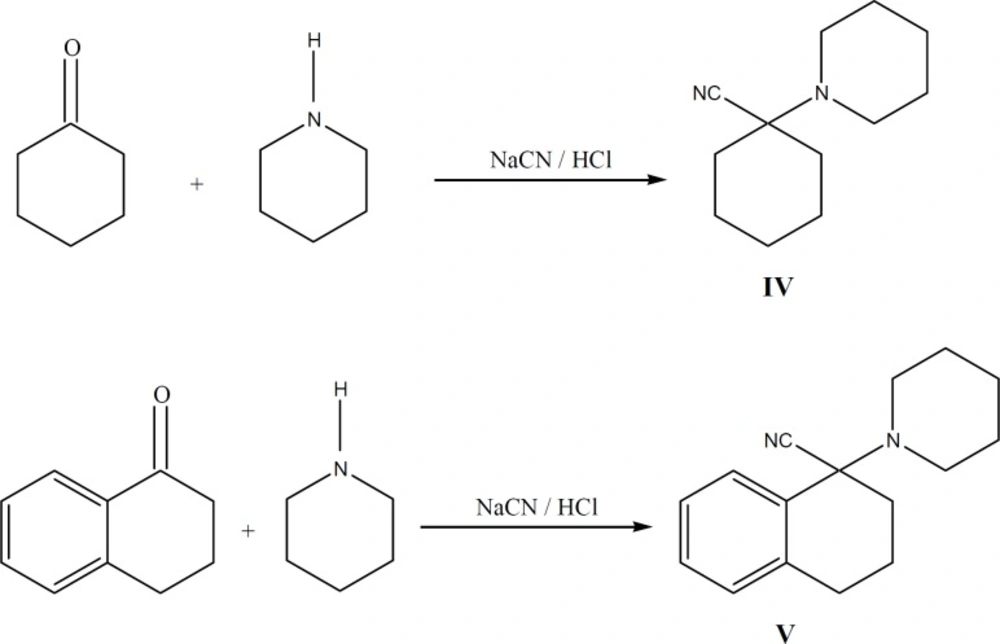
Synthesis of intermediates IV and V
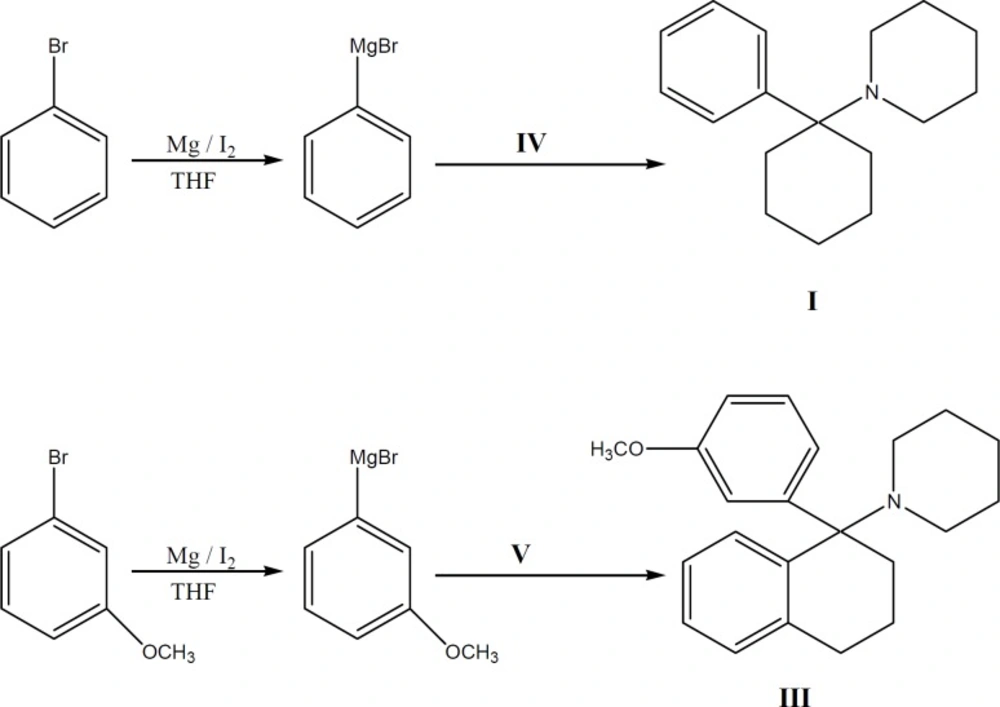
Synthesis of compoun dsIandIII

Mean tail immersion latency (s) in animals receiving ketamine, PCP and PCP-OCH3-tetralyl hydrochloride in saline (control). The tail immersion test was conducted 2, 5, 10, 15, 20, 25, 30, 35 and 40 min after drugs injection. Each point represents the mean ± SEM of tail immersion latencies (s) in 7-9 animals

Comparison of the acute and chronic formalin pain in ketamine, PCP and PCP-OCH3-tetralyl hydrochloride animal groups compared with respect to control. Bars show the mean ± SEM of pain score. As indicated, administration of the drugs had no effect on acute pain (phase I), but the initial and late phases of chronic pain could be significantly reduced following the administration of PCP and especially ketamine. n = 12, * and $ (p < 0.05) show the difference with control and ketamine groups respectively
Recently, many analogues of phencyclidine have been synthesized (7-18) and their pharmacological activities have been studied. As part of our efforts to reach selective, non-competitive antagonists at the PCP binding site on NMDA receptor complex, we have prepared 1-[1-(3-methoxyphenyl) (tetralyl)] piperidine, (PCP-OCH3-tetralyl, III, Figure 1), as an analogue of PCP containing a methoxy group on the aromatic ring (m-position) and a phenyl group with cyclohexane ring (a conjugated cyclic ketone, 1-tetralone). We examined its analgesic effects on rats by tail immersion (as a model of acute thermal pain) and formalin (as a model of acute chemical and chronic pain) tests. The results have been compared with PCP and ketamine. It was anticipated that incorporation of methoxy group on the aromatic ring of the molecule will produce pronounced effect on electron distribution and dipole moments, due to high electron donating character of this group (7). Also it was anticipated that the incorporation of an extra aromatic and flat phenyl group on cyclohexane ring (a conjugated cyclic ketone, 1-tetralone) is able to reduce the conversion of the conformed isomers of the drug (19-21).
Experimental
Materials
1-Tetralone [1, 2, 3, 4-tetrahydro-1-naphthalenone], Cyclohexanone, Piperidine, bromo benzene, magnesium turning, diethyl ether, 3-bromo anizole and all other chemicals, were supplied from Merck Chemical Co. (Darmstadt, Germany). Melting points (uncorrected) were determined using a digital electrothermal melting point apparatus (model 9100, Electrothermal Engineering Ltd., Essex, UK). 1H and 13C NMR spectra were recorded on a Bruker 300 MHz (model AMX, Karlsruhe, Germany) spectrometer (internal reference: TMS). IR spectra were recorded on a Thermo Nicolet FT-IR (model Nexus-870, Nicolet Instrument Corp, Madison, Wisconsin, U.S.A.) spectrometer. Mass spectra were recorded on an Agilent Technologies 5973, Mass Selective Detector (MSD) spectrometer (Wilmington, USA). Chromatographic column separations were performed over Acros silica gel (No.7631-86-9 particle size 35-70 micrometer, Geel, Belgium). Adult female Wistar rats (Pasteur Institute, Tehran, Iran), weighing 250 -300 g were used for pharmacological testing.
Methods
Synthesis of compounds (Figure 1, 2)
(1-(1-phenylcyclohexyl) piperidine (PCP) I
This compound was prepared according to reported method (22) from 1-piperidinocy-clohexanecarbonitrile (IV) and phenyl magnesium bromide. The hydrochloride salt was prepared using 2-propanol and HCl and was recrystallized from 2-propanol (22).
1-Piperidinotetralylcarbonitrile V
To a solution, containing 0.582 g (0.0068 mol) of piperidine in 0.253 g HCl (37%) and 1.36 g cold water, 1 g (0.0068 mol) 1, 2, 3, 4-tetrahydro-1-naphtalenone (1-tetralone) was added. Then 0.465 g KCN in 1.02 mL water, 50 mL ethanol and 0.1 g tetra-n-buthylammonium bromide (0.0003 mol) were added and stirred in ambient temperature (25˚ C). The progress of reaction was controlled by TLC (7:3 ethyl acetate: n-Hexane). After one week no additional progress was observed and so the reaction was performed with chloroform (75 mL, 3 times). Then organic layer was separated, dried and concentrated. The oily residue obtained, was passed through a silica gel column using ethyl acetate: hexane (7:3) as the eluent to afford 1.13 g of V (69 % yield).
IR (KBr): 3066, 2941, 2560, 1454, 1436, 1324, 1287, 1225, 764 cm-1.
1H NMR (CDCl3) δ (ppm): 1.56 (6H, b, β and γ H of piperidine ring), 1.68 (2H, b, β H of cyclohexane ring), 1.85 (2H, b, α H of cyclohexane ring), 2.13 ( 4H, b, α H of piperidine ring), 2.74 ( 2H, b, γ H of cyclohexane ring), 6.82-6.95 (8H, m, ArH).
13C NMR (CDCl3) δ (ppm): 25.4, 26.2, 26.8, 31, 37.9, 46.7, 52.7, 117.7, 125.5, 128.1 and 139.2.
MS: m/z (regulatory intensity): 240 [M]+ (76), 241 [M+ H]+(15).
1-[1-(3-methoxyphenyl) (tetralyl)] Piperidine III
A solution containing 4 g (0.016 mol) of nitrile compound (V) in 10 mL of dry THF was added to a refluxing solution of (3-methoxylphenyl) magnesium bromide (Grignard reagent) (prepared from 24.77 g 3-bromoanisole and 3.075 g of Mg in 17 mL of dry ether), refluxed for 5 additional h in 65-67 oC, left overnight at ambient temperature (25 oC) and then poured into ice-NH4Cl. The organic layer was separated and washed with water and the base was neutralized with 10% H2SO4, washed with 20% NaOH, re-extracted with n-Hexane, dried and concentrated. The obtained oily residue was obtained, which was passed through a silica gel column using ethyl acetate: hexane (7:3) as the eluent to afford 2.28 g of III (42 % yield).
The hydrochloride salt of III was prepared using 2-propanol and HCl and was recrystallized from 2-propanol as an oily compound.
IR (KBr): 3066, 2941, 1602, 1483, 1454, 1436, 1324, 1287, 1225, 764 cm-1.
1H NMR (CDCl3) δ (ppm): 1.51 (6H, b, β and γ H of piperidine ring), 1.62 (2H, b, β H of cyclohexane ring), 1.95 (4H, b, α H of cyclohexane ring), 2.24 (4H, b, α H of piperidine ring), 2.85 (2H, b, γ H of cyclohexane ring), 3.73 (3H, S, -OCH3), 6.93-7.01 (8H, m, ArH).
13C NMR (CDCl3) δ (ppm): 26.2, 27.5, 31.8, 44.8, 47.4, 56, 63, 111.6, 114, 120.2, 120.7, 125.8, 126.2, 128.8, 130, 139.3, 142.8, 144, 162.5.
MS: m/z (regulatory intensity): 321 [M]+ (100), 322 [M+ H]+(7).
Pharmacological methods
Adult female Wistar rats (Pasteur Institute, Tehran), weighing 250-300 g at the begining of the experiment were randomly housed; three to four per cage, in a temperature-controlled colony room under light/dark cycle. Animals were given free access to water and standard laboratory rat chow (Pars Company, Tehran, Iran). All behavioral experiments were carried out between 11 a.m. and 4 p.m. under normal room light at 25 °C. All animals were injected by one of the investigator and evaluated by another. This study was carried out in accordance guidelines the policies provided in the guide for the care and use of Laboratory Animals (NIH) and those of the Research Council of Shahed University of Medical Sciences, Tehran, Iran.
Tail immersion test
Acute thermal pain is modeled by the tail immersion test (23-29). Two, 5, 10, 15, 20, 25, 30, 35 and 40 min after an intraperitoneal (IP) injection of drugs (6 mg/kg (8, 9, 15) which is under LD50 dosage limit of the drugs (9) ) (I, II, III) or an equivalent volume of saline (control), the rats (n = 7-9 in each group) were housed in an animal restrainer. Then, the terminal 5 cm of their tails were first submerged into room temperature water (22~24 °C) to check for an aversion to water and afterwards immersed in 52 °C water. The reaction time between immersing the tail and its removal out of the heated water was measured. A cut-off latency of 20 sec was employed to avoid damaging the tail.
Formalin test
Formalin test introduced by Dubuisson and Dennis (30) was used in our experiments. In this method, formaldehyde solution (50 μl, 2.5%) was injected subcutaneously into the plantar surface of hind paw. The animal was then placed in a plexiglass chamber (30×30×30 cm3) with a mirror at 45o angle underneath, for accurate observation. In the treatment groups, the drugs (ketamine, PCP and PCP-CH3-tetralyl (III)) were administrated intraperitoneally 30 min prior to the formaldehyde injection. Prior to experiment, all animals were brought to the test chamber 5 times with 5 min interval in order to adapt them to the environment. The behavioral pain reactions due to formalin injection were detected and recorded for 1 h. The first 15 min, post-formalin injection is known as the early (I) or acute phase, and the period between 15-60 min is considered the second (II) or chronic phase. However, the chronic phase could be divided to initial (15-40 min) and late (40-60 min) periods.
Statistical analysis
All data are expressed as means ± SEM. The mean latency of tail withdrawal (s) in tail immersion test and pain scores of formalin test in different groups was compared using one-way ANOVA followed by Tukey’s post-hoc test. We considered the probability of p < 0.05 as a significant difference.
Results
Chemistry
Phencyclidine (I), and 1-[1-(3-methylphenyl) (tetralyl) piperidine (III) were synthesized by reaction of substituted Grignard reagents and carbonitrile compounds (IV, V). To obtain higher electron distribution and dipole moment properties (7), a methoxy group was substituted on the aromatic ring of the molecule (III) and to decrease the conversion of conformation isomers of the drug as of our previous results in tetraline series of PCP (9, 20, 21), we synthesized a new analogue of PCP with two additional groups on phenyl and cyclohexane rings of the molecule (III). Known procedures were applied for the synthesis of compounds I and IV with the appropriate modifications described previously (19, 22).
Bromobenzene and its m-methoxy (II) derivative were reacted with magnesium to form Grignard reagents, which were then reacted with appropriate piperidinocyclohexanecarbonitrile (IV) and piperidinotetralylcarbonitrile (V). Reaction between the Grignard reagents and the carbonitriles were slow and incomplete. To overcome this barrier, molar ratio of Grignard reagents to carbonitriles was increased (19, 31).
Spectroscopic data (IR, 1H and 13C NMR, Mass) confirmed the structure of compounds III and V. The melting points of known compounds could also confirm their identity. The purity of each compound was checked by TLC using ethyl acetate: n-hexane as the eluent.
Pharmacology
General consideration
Mortality, morbidity, irritability and other side effects could be associated with drugs administration. However, comparison of the motor coordination index (was measured by Rota-rod apparatus, Harvard, UK) indicated no significant differences between control and treatment rats.
The analgesic activity of PCP (I), ketamine (II) and 1-[1-(3-methoxyphenyl) (tetralyl) piperidine (III) hydrochlorides with tail immersion and formalin test
Intraperitoneal injection of PCP (I), Ketamine (II) and PCP-OCH3-tetralyl (III) hydrochloride (6 mg/kg) which were dissolved in saline, produced a significant analgesic effect in tail immersion test. Comparison of the analgesic effect of ketamine, PCP and PCP-OCH3-tetralyl revealed a pronounced anti-nociception in 2-25 min after ketamine injection, but this analgesic effect remained for 40 min following PCP-OCH3-tetralyl application (Figure 4). However, the data from formalin test showed that the chronic anti-nociception effect of ketamine was higher than that of PCP and PCP-OCH3-tetralyl which have nearly the same analgesic effect (Figure 5). The difference in the tail immersion latencies and pain scores were evaluated using analysis of variance method (ANOVA).
Therefore it seems, strong electron donating properties of the methoxy group on meta position of phenyl ring and the incorporation of an extra aromatic and flat phenyl group with cyclohexane ring (a conjugated cyclic ketone, 1-tetralone) facilitated higher binding to NMDA receptor complex which could increase tail immersion latencies in comparison with ketamine and PCP with higher half life.
Discussion
Electrophysiologic and binding studies revealed that when the channels are in the open or activated state, various antagonists of NMDA receptors, including phencyclidine, ketamine and MK-801 primarily binds to PCP-site (9, 32). Previous studies suggest that ketamine may interact with the NMDA receptor at two potentially distinct sites. The first one is one located within channel pore and the second one is associated with hydrophobic domain of the protein. The binding of the agonist to the receptor is assumed to modify the binding of ketamine to both sites (33).
In the present study a new derivative of PCP (III) and its carbonitrile intermediate having changes in substitutions in its phenyl and cyclohexane rings was synthesized. Stronger analgesic effects of some of our synthesized derivatives of PCP with methyl, methoxy, hydroxyl groups on phenyl and cyclohexane rings was reported (8, 9, 15, 19) and higher electron distribution and dipole moments of methoxy group (7) as well as decreasing the conversion of conformation isomers of the drug were shown (9, 20, 21). Furthermore, due to our previous results in tetraline series of PCP (9), we synthesized a new analogue of PCP with two additional groups on phenyl and cyclohexane rings of the molecule (III) and we studied its analgesic effects.
Comparison of the tail immersion and formalin tests data indicated that PCP-OCH3-tetralyl can diminish thermal but not chemical (formalin) acute pain. It demonstrated that perhaps different mechanisms are involved in thermal and chemical acute pain for such different responses. However, the long lasting effect for III compared to II, could be related to higher half-life of III compared to II. Perhaps, the impermeability of blood brain barrier (BBB) to PCP-OCH3-tetralyl could explain the non significancy in analgesic effect of III with regard to the marked analgesic effect of ketamine and PCP in initial and late phases of formalin-induced chronic pain (phase II). Chronic formalin pain (phase II) mediated by inflammatory mediators and CNS activation (34).
