Materials
Olanzapine was obtained as a gift sample form Okasa Pharma Pvt. Ltd., (Satara, India) and from Cipla Ltd. (Kurkumbh, India), 2-hydroxypropyl-β-cyclodextrin was generously donated by Signet Chemical corporation, (Mumbai, India), Aspartame was obtained as a gift sample from Sun and Kingly Pharma Pvt. Ltd. (Satara, India), Tulsion 339 was a gift from Thermax India Ltd. (Pune, India), Indion 414 was kindly donated by Ion Exchange India Ltd. (Mumbai, India) and All other excipients ethanol, avicel PH102, mannitol, SSG, croscarmellose sodium, crospovidone, vanilin dry flavor, aerosil and Mg stearate were procured from Rajesh Chemicals (Mumbai, India). All reagents and solvents were of analytical grade.
Methods
Phase solubility analysis for olanzapine
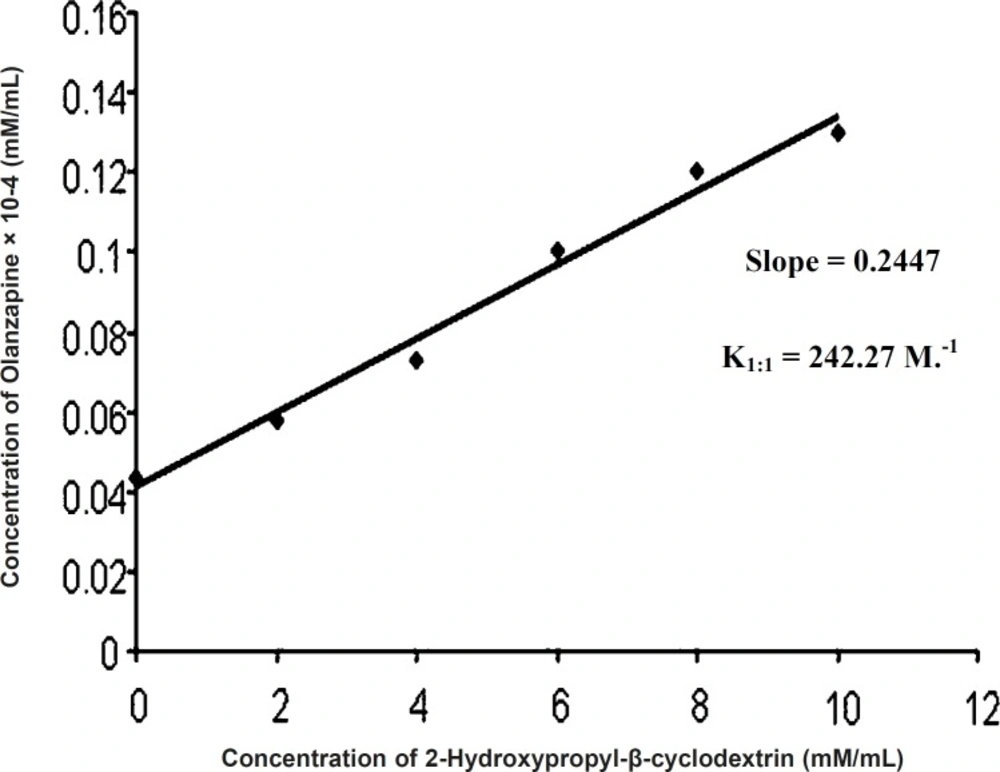
Phase solubility studies were performed to determine stoichiometric proportions. This data was used to determine stability constant of complex. For this purpose, the stock solution of 12 mM of 2-hydroxypropyl-β-cyclodextrin was prepared using distilled water. This stock solution was diluted with distilled water to give molar solutions in the range of 0 mM to 12 mM of 2-hydroxypropyl-β-cyclodextrin. 5 mL of each molar solution was filled in a screw capped vials and the excess quantity of olanzapine was added to each vial separately. The vials were kept for shaking at ambient temperature for 24 h using a lab shaker (Remi). After the complete equilibration, the supernatant solutions were collected carefully and filtered using Whatman filter paper (No. 41). The concentration of olanzapine in filtered solutions was determined using UV visible spectrophotometer (UV-1700 Schimadzu spectrophotometer, Tokyo, Japan) at 253.5 nm. No change in λ max of drug was observed after complexing with cyclodextrins. The graph was plotted against drug concentration vs. concentration of 2-hydroxypropyl-β-cyclodextrin.
The blanks were prepared using the same concentration of 2-hydroxypropyl-β-cyclodextrin in distilled water so as to cancel out any absorbances that may be exhibited by 2-hydroxypropyl-β-cyclodextrin molecules (
11,
12).
Concentration = 1/Slope × Absorbance
The stability constant of the olanzapine 2-hydroxypropyl-β-cyclodextrin inclusion complex was determined from the slope of linear portion of the curves and intrinsic solubility of olanzapine in an aqueous solution using equation:
Stability constant = Slope/Intercept (1-Slope)
Preparation of inclusion complex
Kneading method
Required quantities of olanzapine and cyclodextrin was weighed accurately in 1:1 molar ratio. A homogenous paste of cyclodextrin and olanzapine was prepared in mortar by adding ethanol in small quantities. An appropriate quantity of ethanol was added to maintain suitable consistency of paste. The paste was kneaded for 1 h and then dried in hot air oven (Singhal scientific) at 45-50°C for 3 h. The dried complex was powdered and passed through sieve No. 60 and stored in airtight containers till further use (
13,
14).
Characterization of Olanzapine-2-hydroxypropyl- β-cyclodextrin inclusion complex
Inclusion complex was characterized and evaluated using following techniques.
Drug content estimation
Olanzapine-2-hydroxypropyl-β-cyclodextrin complex, equivalent to 10 mg of drug, was accurately weighed and added in to 100 mL volumetric flask. To this, 100 mL ethanol was added.
This solution was stirred for 60 min, till the entire drug leached out. The solution was filtered and 1 mL was withdrawn from this solution and added in to 10 mL volumetric flask and volume was made to 10 mL (10 μg/mL) with phosphate buffer pH 6.8. Drug content was estimated UV spectrophotometrically at 253.5 nm, using phosphate buffer pH 6.8 as blank.
Saturation solubility studies
Saturation solubility study was performed according to method reported by Higuchi and Connors (
11). Excess quantities of inclusion complex were added to 25 mL distilled water in a stoppered conical flasks and mixtures were shaken for 24 h in rotary flask shaker.
After shaking, to achieve equilibrium, 2 mL aliquots were withdrawn at 1 h intervals and filtered through Whatman filter paper No. 41. The filtrate was analyzed spectrophotometrically at 253.5 nm. Shaking was continued until three consecutive readings were the same (
13).
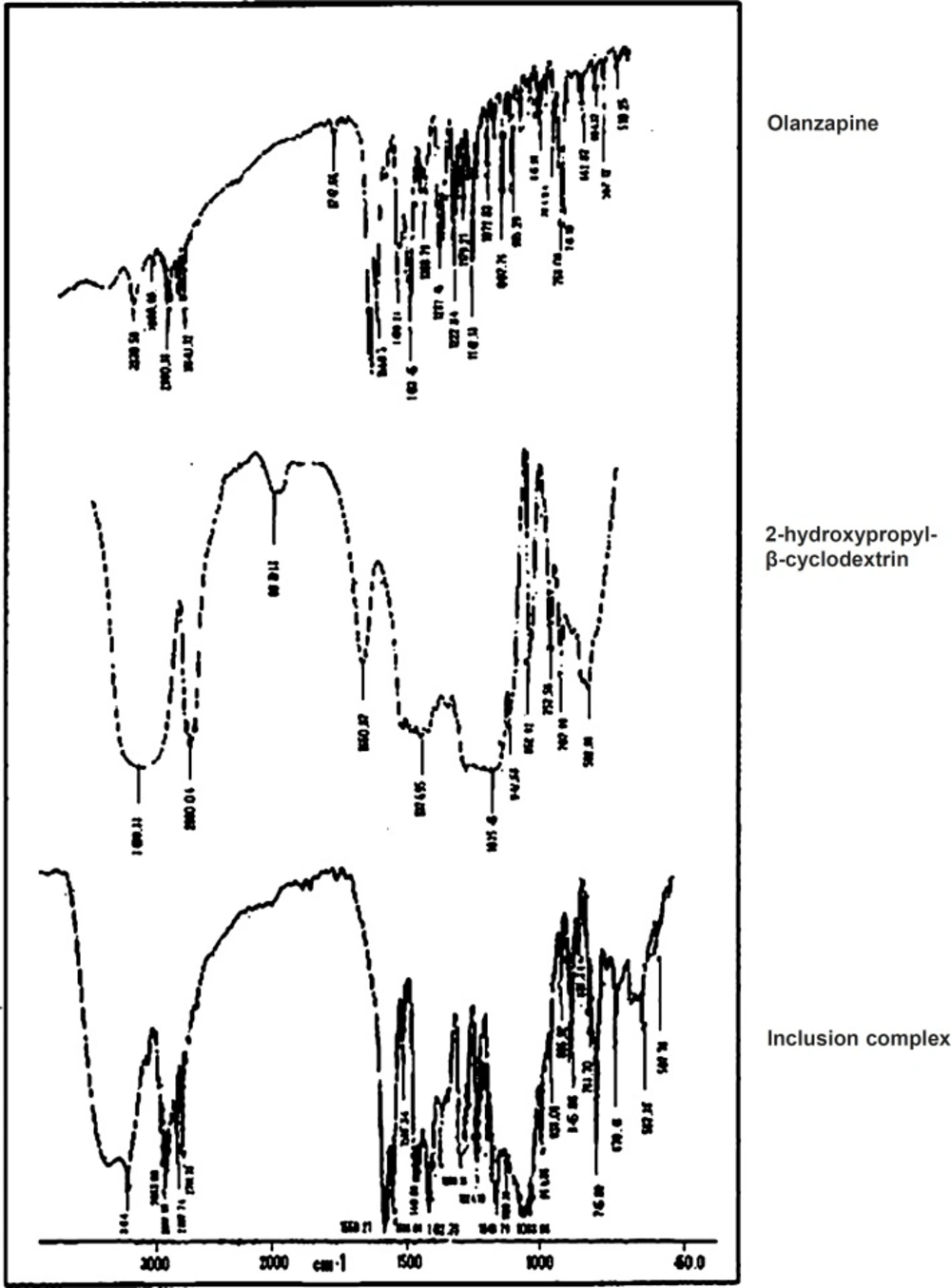
IR spectral analysis
Infrared spectra of pure olanzapine, 2-hydroxypropyl-β-cyclodextrin and inclusion compelx were recorded by KBr method using fourier transform infrared spectrophotometer (spectrum one, by Perkin-Elmer, USA). A baseline correction was made by using dried potassium bromide and then spectras of dried mixtures of olanzapine, 2-hydroxypropyl-β-cyclodextrin and inclusion complex with potassium bromide were recorded. Scanning was done from 450 -4000 cm-1.
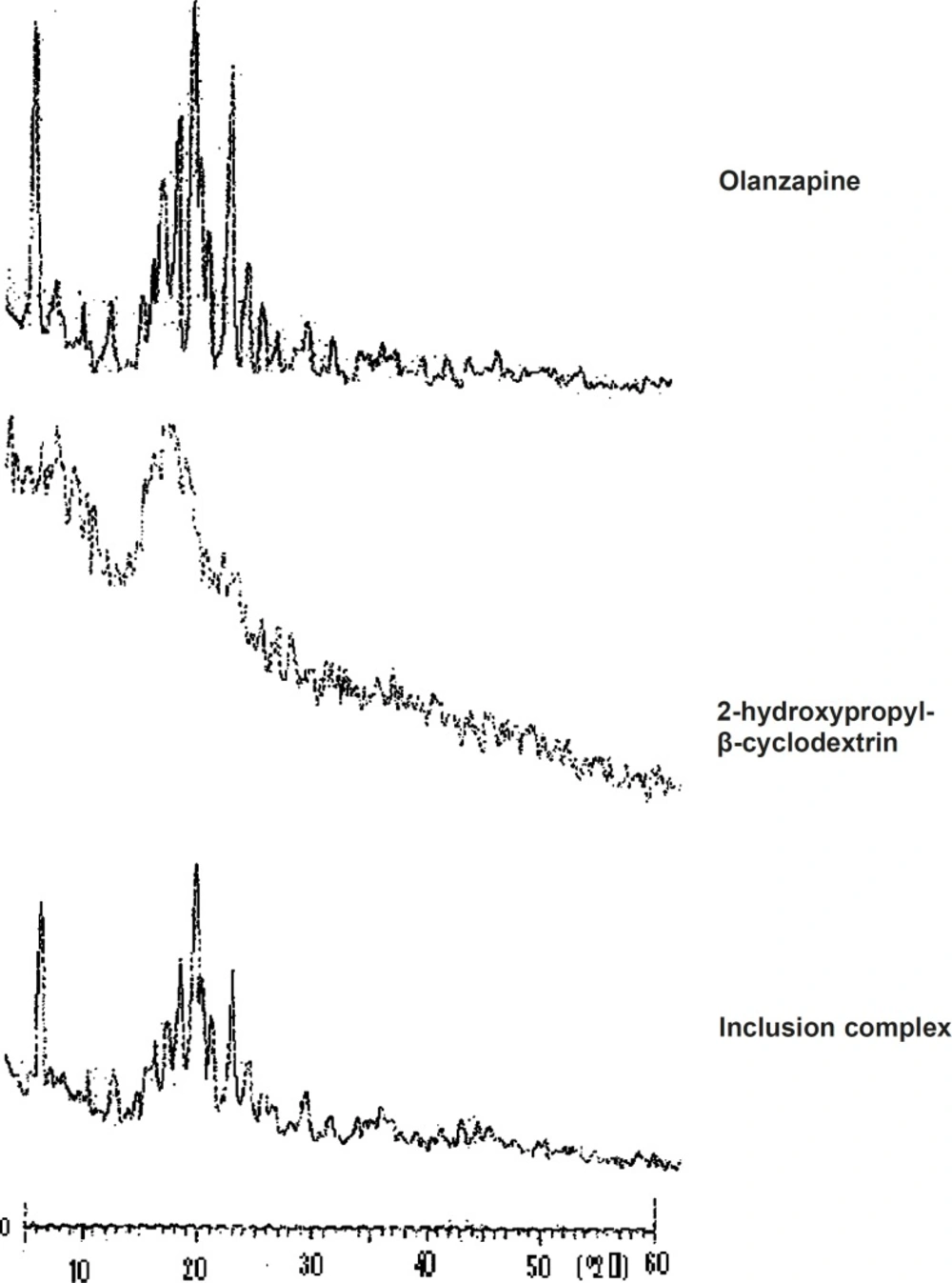
X-ray diffractometery (XRD)
X-ray diffraction patterns of pure olanzapine, 2-hydroxypropyl-β-cyclodextrin and inclusion complex were recorded using (Philips-PW3710, Holland) X-ray diffractometer with a copper target, voltage 40 Kv, current 30 MA at a scanning speed of 0.30 °C /min.
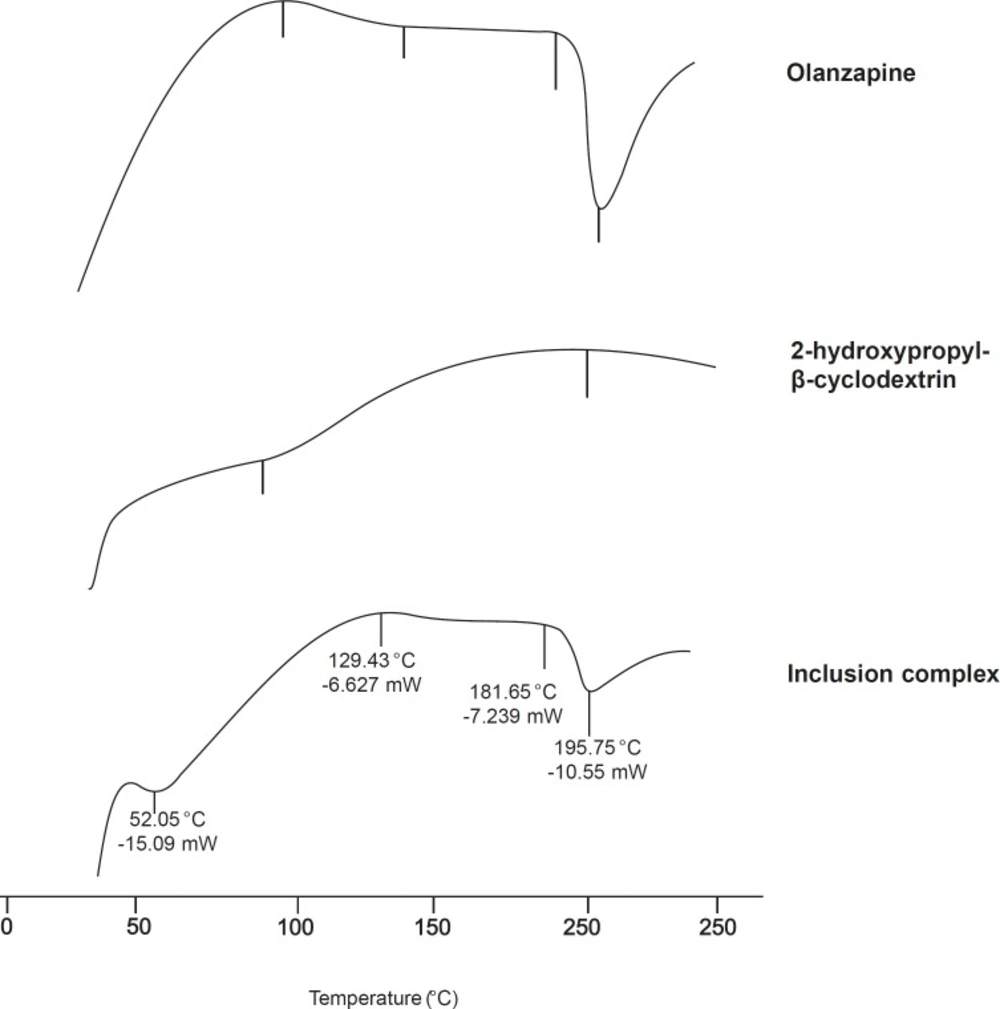
Differential scanning calorimetry (DSC)
Thermograms of pure Olanzapine, 2-hydroxypropyl-β- cyclodextrin and inclusion complex were recorded. (TA instruments Inc, SDT 2960, USA). About 5 mg of samples were sealed in aluminum pans and heated at a rate of 20 °C/min. from 20-250 °C under nitrogen atmosphere of 100 mL/min flow rate (
15).
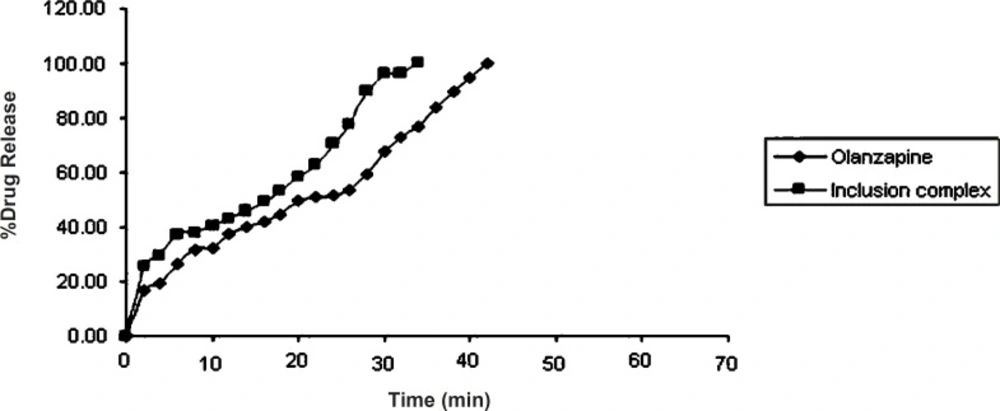
Dissolution study of olanzapine and its inclusion complex
In-vitro dissolution of olanzapine (10 mg) and its inclusion complex equivalent to 10 mg of olanzapine was studied using Veego scientific, USP tablet dissolution apparatus type II. The dissolution was carried out in 900 mL phosphate buffer pH 6.8. at 37 ± 0.5 °C, at 50 rpm. 10 mL aliquots were withdrawn at specific time interval and filtered using Whatman filter paper No. 41. Absorbance of the filtered solution was checked UV spectrophotometrically at 253.5 nm and the drug content was determined. Sink conditions were maintained throughout the study. All studies were carried out in triplicate (
3).
Formulation and evaluation of tablets
Formulation of tablets
ODTs of olanzapine were prepared by direct compression method according to the formula given in
Table 1. Various superdisinterants with different concentrations were used such as SSG, croscarmellose sodium, crospovidone, tulsion 339 and indion 414. olanzapine-2-hydroxypropyl-β-cyclodextrin inclusion complex and Avicel PH102 were passed through sieve No. 60 and mixed homogeneously for 15 min (blend I). Mannitol, superdisintegrant, aspartame, vanillin and aerosil were passed through sieve No. 40 and mixed homogeneously for 10 min (called as blend II). Blend II was mixed with blend I and mixing was continued for 1 h.
| Name of ingredients Qty.in mg. | Control | A1 | A2 | A3 | B1 | B2 | B3 | C1 | C2 | C3 | D1 | D2 | D3 | E1 | E2 | E3 |
|---|
| Inclusion complex | 54.80 | 54.80 | 54.80 | 54.80 | 54.80 | 54.80 | 54.80 | 54.80 | 54.80 | 54.80 | 54.80 | 54.80 | 54.80 | 54.80 | 54.80 | 54.80 |
| Superdisintegrant | - | 14 | 15.75 | 17.5 | 5.25 | 7 | 8.75 | - | - | - | 5.25 | 7 | 8.75 | 5.25 | 7 | 8.75 |
| Avicel PH102 | 75.45 | 61.45 | 59.7 | 57.95 | 70.2 | 68.45 | 66.7 | 70.2 | 68.45 | 66.7 | 70.2 | 68.45 | 66.7 | 70.2 | 68.45 | 66.7 |
| Aspartame | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 |
| Mannitol | 41 | 41 | 41 | 41 | 41 | 41 | 41 | 41 | 41 | 41 | 41 | 41 | 41 | 41 | 41 | 41 |
| Vanilin dry flavor | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | | 1 |
| Aerosil | 0.875 | 0.875 | 0.875 | 0.875 | 0.875 | 0.875 | 0.875 | 0.875 | 0.875 | 0.875 | 0.875 | 0.875 | 0.875 | 0.875 | 0.875 | 0.875 |
| Mg.stearate | 0.875 | 0.875 | 0.875 | 0.875 | 0.875 | 0.875 | 0.875 | 0.875 | 0.875 | 0.875 | 0.875 | 0.875 | 0.875 | 0.875 | 0.875 | 0.875 |
| Tablet weight | 175 | 175 | 175 | 175 | 175 | 175 | 175 | 175 | 175 | 175 | 175 | 175 | 175 | 175 | 175 | 175 |
Finally, Magnesium stearate was added and mixed for 1 min. This blend was compressed using 8 mm size flat faced punch on (Cadmach) single punch compression machine.
Control formulation was formulated without adding superdisintegrant. Mannitol improves patient compliance by imparting cool sensation and sweet mild taste. It flows well and improves flow properties of other materials (
16). Aspartame was used as sweetener. It enhanced flavor systems and also used as taste masking agent. The approximate sweetening power is 180-200 times to that sucrose. Stability of aspartame can be enhanced by addition of cyclodextrin, vanilin dry flavor was used as a flavoring agent (
17). It imparts a characteristic taste and odor of natural vanilla. Aerosil was used as glidant, magnesium stearate was used as lubricant (
17).
Evaluation of tablets
Diameter
It was measured by digital vernier caliper. It is expressed in mm.
Thickness
It was determined by using digital vernier caliper. It is expressed in mm.
Crushing strength
Tablet crushing load, which is the force required to break a tablet by compression in radial (diametrical) direction was measured by using tablet hardness tester. Crushing strength for crushing (T) was calculated using the following formula.
T = 2F/ΠDT F = Crushing load
D = Diameter
T = Thickness
Hardness
The hardness of the tablets was determined using Monsanto hardness tester. It is expressed in kg/cm
2 (
18).
Friability
Friability of the tablets was determined using Roche friabilator. It is expressed in % (
18).
Disintegration time
It was measured by two methods
I) Using USP disintegration test apparatus.
II) Using modified disintegration test apparatus.
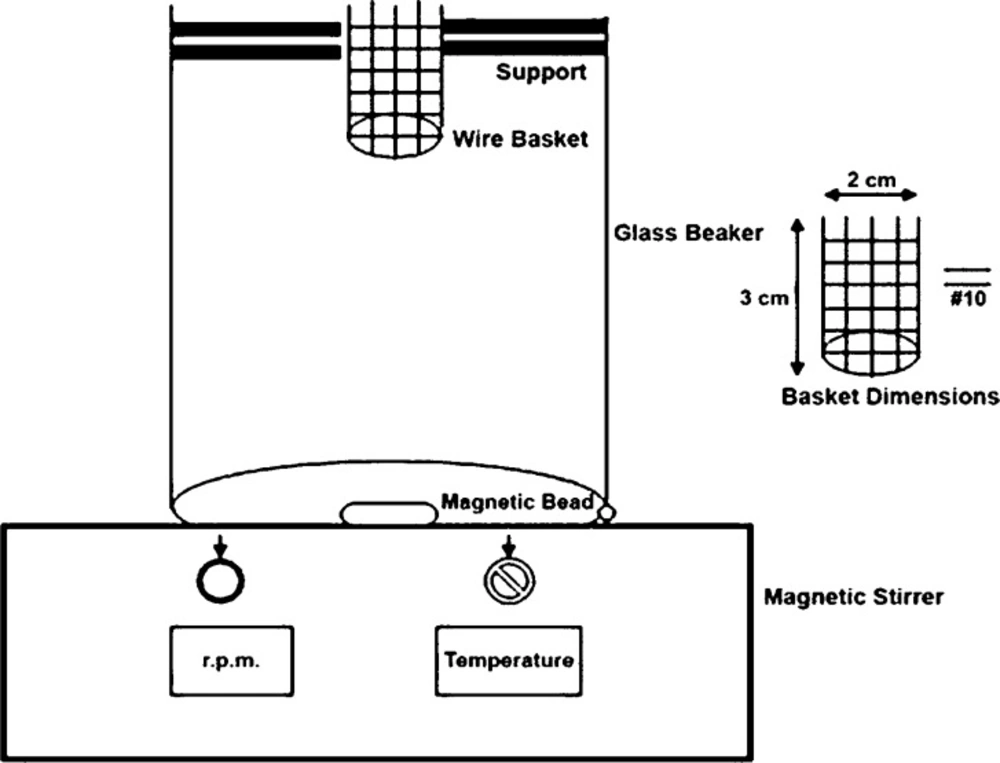
In-vitro disintegration time for ODTs was determined using USP and modified disintegration test apparatus with pH 6.8 phosphate buffer as disintegrating medium (
19). During this study we made an attempt to develop a more suitable apparatus for ODT because many reports indicated the unsuitability of conventional disintegration test apparatus for ODT (
20,
21,
22,
23). Briefly, (
Figure 1) the apparatus consisted of a glass beaker of 1000 mL capacity with the wire basket positioned in the beaker with the help of a support in a way that when the beaker contained 900 mL of phosphate buffer pH 6.8 as a disintegration medium, the basket had only 6 mL of it. A magnetic bead was placed at the bottom of a beaker and temperature was maintained at 37 ± 2 °C. Disintegration time was determined at 50 rpm (
24). The results of two methods were compared.
Modified disintegration test apparatus
Weight variation
20 tablets were selected at random and an average weight was determined (Electronic balance Adventure Ohaus, USA). Not more than two of individual weights deviated from the average weight by ± 7.5% (
18).
Uniformity of content
Drug content from the tablets was determined by taking tablets from each formulation. Twenty tablets from each formulation were accurately weighed and powdered. Powder equivalent to 10 mg of drug was weighed and added into 100 mL volumetric flask. Then it was dissolved in 50 mL ethanol. The volume of solution was made to 100 mL (100 μg/mL). From this solution 1 mL was withdrawn and added into 10 mL volumetric flask and finally volume was made to 10 mL with phosphate buffer pH 6.8 (10 μg/mL), then solution was filtered with Whatman filter paper No. 41 and absorbance of the resulting solution was measured at 253.5 nm using UV spectrophotometer.
Wetting time and water absorption ratio
Wetting time is closely related to the inner structure of tablets and hydrophilicity of the excipients. According to the following equation proposed by washburn E.W (
25), the water penetration rate into the powder bed is proportional to the pore radius and is affected by the hydrophilicity of the powders.
dl / dt = r γ cosθ / (4 ηl)
l = length of penetration
r = capillary radius
γ = surface tension
η = liquid viscosity
t = time
θ = contact angle
A piece of a tissue paper folded twice was placed in a small petri plate (internal diameter 6.5 cm) containing 6 mL of water. A tablet was placed on the paper and time for complete wetting of the tablet was measured in seconds. The method was slightly modified by maintaining water at 37 ˚C. The same procedure was followed for determining the water absorption ratio. The wetted tablet was weighed and water absorption ratio, R, was determined according to the following equation:
R = { (Wa - Wb) / Wb } × 100
Where, Wa and Wb were the weights of the tablet after and before study (
24,
25).
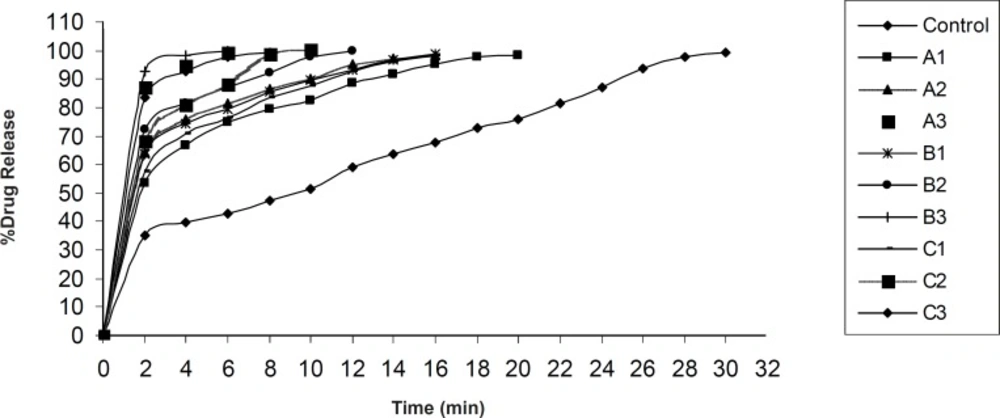
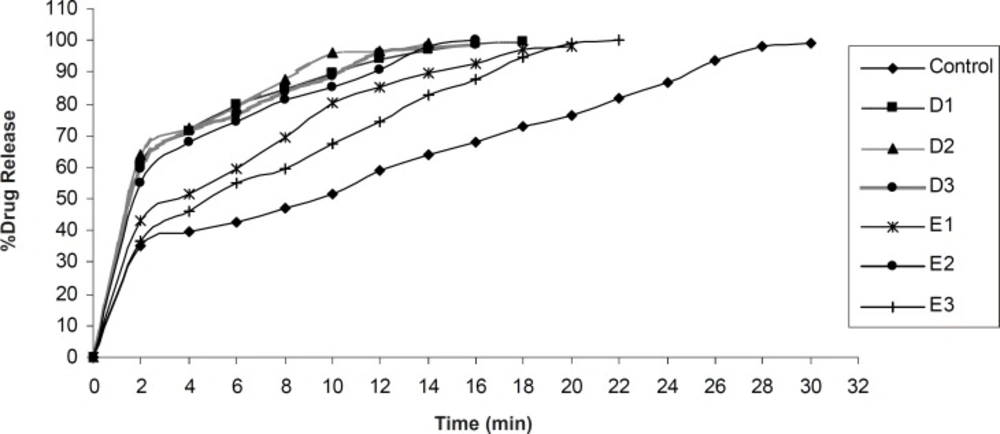
In-vitro dissolution study
In-vitro dissolution of olanzapine ODTs was studied using Veego scientific, USP tablet dissolution apparatus type II. The dissolution was carried out in 900 mL phosphate buffer pH 6.8. at 37 ± 0.5 ˚C, at 50 rpm. 10 mL aliquots were withdrawn at specific time interval and filtered using Whatman filter paper No. 41. Absorbance of the filtered solution was checked UV spectrophotometrically at 253.5 nm and the drug content was determined. Sink conditions were maintained throughout the study. All studies were carried out in triplicate (
3).
Statistical analysis
One way ANOVA with Students Paired ‘t’ test was applied to compare the dissolution rate of pure olanzapine with its inclusion complex and to compare the disintegration time between USP disintegration test apparatus and Modified disintegration test apparatus. One way ANOVA with Dunnett’s post test was applied to find out significant difference in time to release 100% olanzapine from different formulations.