Chemistry
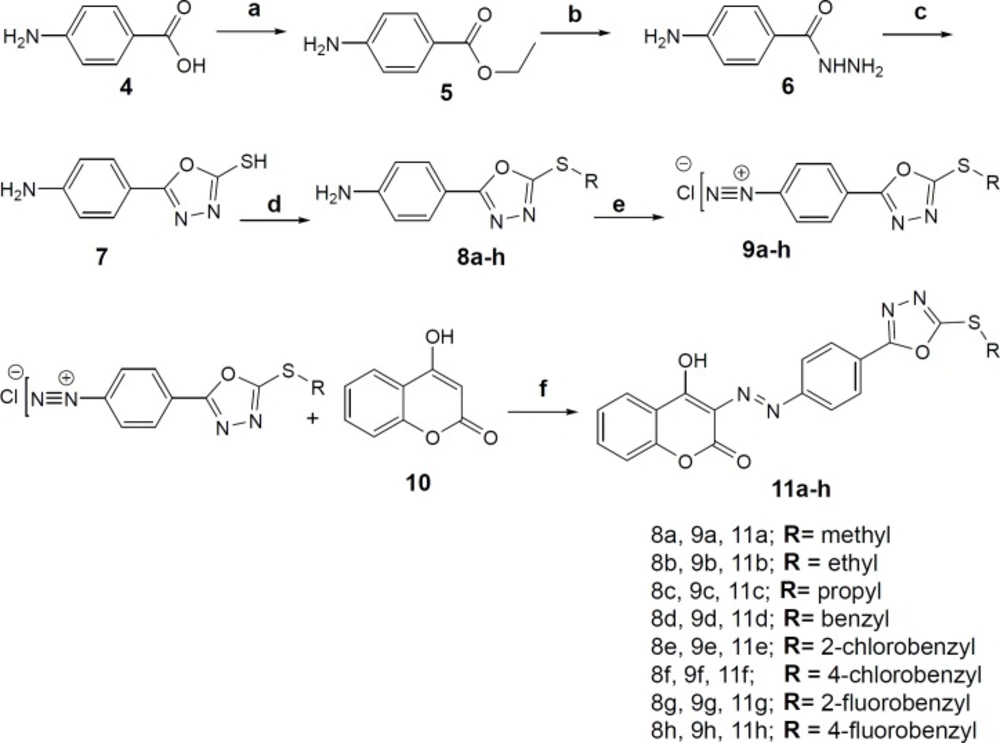
3-((4-(5-(substitutedaryl/alkyl)-1,3,4-oxadiazol-2-yl) phenyl) diazenyl)-4-hydroxy-2
H-chromen-2-onederivatives were synthesized according to the
scheme 1. First, Ethyl 4-aminobenzoate was prepared from 4-aminobenzoic acid (
4) and ethanol in a routine Fischer esterification reaction under sulfuric acid catalysis. Ethyl 4-aminobenzoate (
5) was converted to 4-aminobenzohydrazide (
6) by refluxing in absolute ethanol and excess of hydrazine hydrate. The 2-mercaptho-1,3,4-oxadiazole ring (
7) was closed on the carbohydrazide intermediate in the presence of carbon disulfide and alcoholic potash according to procedure described in references (
26,
27). S-alkylation/benzylation reaction was performed with alkyl/ substituted benzyl halides in methanol as the solvent and 10% sodium hydroxide solution to afford derivatives
8a-h. The moderate basicity of the reaction medium is essential to gain the mono S-alkyl/benzyl product rather than both S- and N-alkyl/benzyl substituted product. The diazonium salts of compounds (
9a-h) were prepared in 6 M hydrochloric acid and 10% sodium nitrite aqueous solution at 0-5 °C. The final derivatives
11a-h were obtained through coupling reaction of the diazonium salts with 4-hydroxycoumarin (
10) in aqueous sodium carbonate solution at neutral pH.
Materials
All reagents purchased from the Aldrich (USA) or Merck (Germany) Chemical Company and were used without further purifications.
General
Melting points (mp) were determined using a Thomas Hoover capillary apparatus (Philadelphia, USA). Infrared spectra were acquired on a Perkin-Elmer 1420 ratio recording spectrometer. A Bruker FT-500 MHz instrument (Brucker Biosciences, USA) was used to acquire 1HNMR; Chloroform-D used as solvent. Coupling constant (J) values are estimated in hertz (Hz) and spin multiples are given as brs (broad singlet), s (singlet), d (double), t (triplet), q (quartet), m (multiplet), and br (broad). The mass spectral measurements were performed on an 6410 Agilent LCMS triple quadrupole mass spectrometer (LCMS) with an electrospray ionization (ESI) interface.
Ethyl 4-aminobenzoate (5)
Yield 74%; White crystalline powder; mp 89.2-91.0 °C; IR (KBr disk): υ (cm-1) 3424- 3348 (NH2), 1682 (C=O); LC-MS (ESI) m/z: 166.1 (M+1, 100).
4-Aminobenzohydrazide (6)
Yield 82%; White crystalline powder; mp 216.8-218.3 °C; IR (KBr disk): υ (cm-1) 3432- 3350 (NH2), 3307-3278 (-CO-NHNH2), 3239 (-CO-NHNH2), 1603 (C=O); LC-MS (ESI) m/z: 152.0 (M+1, 100).
5-(4-Aminophenyl)-1,3,4-oxadiazole-2-thiol (7)
To a solution of compound 6 (6 mmol) and potassium hydroxide (1.23 g) in absolute ethanol (50 mL), carbon disulfide (8.2 mL) was added in an ice bath. The mixture was refluxed at 50-60 °C for 4 hours. Then, it was cooled to room temperature and the solvent was evaporated to dryness. The crude suspended in 7 mL water and acidified with 3 M hydrochloric acid until pH 4.5. The yellow precipitate was filtered and washed with water. Yield 73%; Yellow powder; mp 197-199 °C; IR (KBr disk): υ (cm-1) 3356- 3440 (NH2); LC-MS (ESI) m/z: 194.0 (M+1, 100).
General procedure for preparation of 4-(5-(substituted thio)-1,3,4-oxadiazol-2-yl) aniline (8a-h)
To a solution of compound 7 (1.55 mmol) in methanol (4 mL) sodium hydroxide (10%, 0.12 mL) was added. The mixture was cooled in an ice bath and substituted benzyl/alkyl halide (1.55 mmol) was added dropwise and stirred at room temperature. After completion of the reaction, the precipitate was filtered, rinsed with water, and crystallized from ethanol.
4-(5-(Methylthio)-1,3,4-oxadiazol-2-yl)aniline (8a)
Yield 87%; Yellow crystalline powder; mp 105.8-107.6 °C; IR (KBr disk): υ (cm-1) 3327- 3425 (NH2); LC-MS (ESI) m/z: 207.9 (M+1, 100).
4-(5-(Ethylthio)-1,3,4-oxadiazol-2-yl)aniline (8b)
Yield 73%; Yellow crystalline powder; mp 80.4-83.2 °C; IR (KBr disk): υ (cm-1) 3374-3444 (NH2); LC-MS (ESI) m/z: 221.9 (M+1, 100).
4-(5-(Propylthio)-1,3,4-oxadiazol-2-yl)aniline (8c)
Yield 67%; Yellow crystalline powder; mp 105.1-106.8 °C; IR (KBr disk): υ (cm-1) 3333-3478 (NH2); LC-MS (ESI) m/z: 236.9 (M+1, 100).
4-(5-(Benzylthio)-1,3,4-oxadiazol-2-yl)aniline (8d)
Yield 61%; Yellow crystalline powder; mp 120.8-124 °C; IR (KBr disk): υ (cm-1) 3359-3493 (NH2); LC-MS (ESI) m/z: 284.9 (M+1, 100).
4-(5-(2-Chlorobenzylthio)-1,3,4-oxadiazol-2-yl)aniline (8e)
Yield 59%; Yellow crystalline powder; mp 117.8-118.9 °C; IR (KBr disk): υ (cm-1) 3337-3399 (NH2); LC-MS (ESI) m/z: 318.9 (M+1, 100).
4-(5-(4-Chlorobenzylthio)-1,3,4-oxadiazol-2-yl) aniline (8f)
Yield 65%; Yellow crystalline powder; mp 141.8-144.3 °C; IR (KBr disk): υ (cm-1) 3349-3417 (NH2); LC-MS (ESI) m/z: 318.9 (M+1, 100).
4-(5-(2-Fluorobenzylthio)-1, 3, 4-oxadiazol-2-yl) aniline (8g)
Yield 55%; Yellow crystalline powder; mp 121.8-123.8 °C; IR (KBr disk): υ (cm-1) 3373-3506 (NH2); LC-MS (ESI) m/z: 302.9 (M+1, 100).
4-(5-(4-Fluorobenzylthio)-1, 3, 4-oxadiazol-2-yl) aniline (8h)
Yield 59%; Yellow crystalline powder; mp 104-106 °C; IR (KBr disk): υ (cm-1) 3353-3422 (NH2); LC-MS (ESI) m/z: 302.9 (M+1, 100).
General procedure for the preparation of (E)-4-hydroxy-3-((4-(5-(substitutedthio)-1, 3, 4-oxadiazol-2-yl) phenyl)diazenyl)-2H-chromen-2-one (11a-h)
To a cooled solution of substituted compounds 8a-h (1 mmol) in 6 M hydrochloric acid (2.7 mL), 0.7 mL cooled solution of 10% sodium nitrite was added dropwise in order to keep the temperature less than 5 °C. Then, the mixture was poured into a solution of 4-hydroxy coumarin (10) (1 mmol) and sodium carbonate (0.05 g) in water (1.85 mL). The pH of the mixture was adjusted to 7 by addition of sodium acetate. The reaction mixture was stirred in an ice bath. After completion of the reaction, the precipitate was filtered, washed with water and crystallized from acetonitrile.
4-Hydroxy-3-((4-(5-(methylthio)-1, 3, 4-oxadiazol-2-yl) phenyl)diazenyl)-2H-chromen-2-one (11a)
Yield 81%; Orange powder; mp 211.6-212.1 °C; IR (KBr disk): υ (cm-1) 1666-1744 (C=O); 1HNMR (CDCl3, 500 MHz): δ (ppm) 2.59 (3H, s, -CH3); 7.32 (1H, d, J= 7.5 Hz, H8) 7.34-7.39 (1H, m, H6), 7.68-7.74 (3H, m, H7 & phenyl H2 & H6), 7.98 (2H, brs, phenyl H3 & H5), 8.09 (1H, d, J= 7.4 Hz, H5); LC-MS (ESI) m/z: 381.2 (M+1, 100). Anal. Calcd. for C18H12N4O4S; C, 56.84; H, 3.18; N, 14.73. Found: C, 56.99; H, 3.32; N, 14.52.
3-((4-(5-(Ethylthio)-1, 3, 4-oxadiazol-2-yl) phenyl)diazenyl)-4-hydroxy-2H-chromen-2-one (11b)
Yield 78%; Orange powder; mp 290-291 °C (decomposed); IR (KBr disk): υ (cm-1) 1760 (C=O); 1HNMR (CDCl3, 500 MHz): δ (ppm) 1.53 (3H, t, J= 7.3 Hz, -CH3), 3.34 (2H, q, J= 7.3 Hz, -CH2), 7.30 (1H, d, J= 8.2 Hz, H8), 7.34 (1H, t, J= 7.5 Hz, H6), 7.70 (1H, t, J= 7.3 Hz, H7), 7.78 (2H, d, J= 8.5 Hz, phenyl H2 & H6), 8.09-8.14 (3H, m, H5 & phenyl H3 & H5); LC-MS (ESI) m/z: 395.0 (M+1, 100). Anal. Calcd. for C19H14N4O4S; C, 57.86; H, 3.58; N, 14.21. Found: C, 57.66; H, 3.41; N,
14.01.
4-Hydroxy-3-((4-(5-(propylthio)-1, 3, 4-oxadiazol-2-yl) phenyl)diazenyl)-2H-chromen-2-one (11c)
Yield 71%; Orange powder; mp 205.5-206.8 °C; IR (KBr disk): υ (cm-1) 1732 (C=O); 1HNMR (CDCl3, 500 MHz): δ (ppm) 1.09 (3H, t, J= 7.3 Hz, -CH3), 1.89 (2H, sixtet, J= 7.3 Hz, CH3-CH2), 3.30 (2H, t, J= 7.2 Hz, CH2-CH2), 7.30 (1H, d, J= 8.3 Hz, H8), 7.34 (1H, t, J= 7.5 Hz, H6), 7.70 (1H, t, J= 8.4 Hz, 7.2, H7), 7.78 (2H, d, J= 8.6 Hz, phenyl H2 & H6), 8.09-8.1 (3H, m, H8 & phenyl H3 & H5); LC-MS (ESI) m/z: 409.2 (M+1, 100). Anal. Calcd. for C20H16N4O4S; C, 58.82; H, 3.95; N, 13.72. Found: 58.61; H, 3.77; N, 13.55.
3-((4-(5-(Benzylthio)-1, 3, 4-oxadiazol-2-yl) phenyl)diazenyl)-4-hydroxy-2H-chromen-2-one (11d)
Yield 75%; Orange powder; mp 243.1-243.8 °C; IR (KBr disk): υ (cm-1) 1737 (C=O); 1HNMR (CDCl3, 500 MHz): δ (ppm) 4.58 (2H, s, -CH2), 7.35-7.39 (5H, m, H6 & H8 & benzyl H3 & H4 & H5), 7.50 (2H, d, J= 7.2 Hz, benzyl H2 & H6), 7.74 (1H, m, H7), 7.82 (2H, d, J= 8.2 Hz, phenyl H2 & H6), 8.13-8.15 (3H, m, H5 & phenyl H3 & H5); LC-MS (ESI) m/z: 284.1 (M+1, 100). Anal. Calcd. for C24H16N4O4S; C, 63.15; H, 3.53; N, 12.27. Found: C, 62.95; H, 3.41; N, 12.57.
3-((4-(5-(2-Chlorobenzylthio)-1, 3, 4-oxadiazol-2-yl) phenyl)diazenyl)-4-hydroxy-2H-chromen-2-one (11e)
Yield 71%; Orange powder; mp 236.8-237.5 °C; IR (KBr disk): υ (cm-1) 1755 (C=O); 1HNMR (CDCl3, 500 MHz): δ (ppm) 4.69 (2H, s, -CH2), 7.30-7.45 (5H, m, H6 & H8 & 2-Cl-benzyl H4 & H5 & H6), 7.67-7.73 (2H, m, H7 & 2-Cl-benzyl H3), 7.82 (2H, m, phenyl H2 & H6), 8.13-8.4 (3H, m, H5 & phenyl H3 & H5), 16.26 (1H, s, -OH); LC-MS (ESI) m/z: 491.2 (M+1, 100). Anal. Calcd. for C24H15ClN4O4S; C, 58.72; H, 3.08; N, 11.41. Found: C, 58.62; H, 3.28; N,
11.25.
3-((4-(5-(4-Chlorobenzylthio)-1, 3, 4-oxadiazol-2-yl) phenyl)diazenyl)-4-hydroxy-2H-chromen-2-one (11f)
Yield 74%; Orange powder; mp 260.8-263.8 °C; IR (KBr disk): υ (cm-1) 1748 (C=O); 1HNMR (CDCl3, 500 MHz): δ (ppm) 4.49 (2H, s, -CH2), 7.30-7.36 (4H, m, H6 & H8 & 4-Cl-benzyl H2 & H6), 7.41 (2H, d, J= 8.5, 4-Cl-benzyl H3 & H5), 7.70 (1H, m, H7), 7.78 (2H, d, J= 10, phenyl H2 & H6), 8.09 (3H, m, H5 & phenyl H3 & H5); LC-MS (ESI) m/z: 514.3 (M+23, 100). Anal. Calcd. for C24H15ClN4O4S; C, 58.72; H, 3.08; N, 11.41. Found: C, 58.98; H, 3.23; N, 11.30.
3-((4-(5-(2-Fluorobenzylthio)-1, 3, 4-oxadiazol-2-yl) phenyl)diazenyl)-4-hydroxy-2H-chromen-2-one (11g)
Yield 58%; Orange powder; mp 240.1-242.2 °C; IR (KBr disk): υ (cm-1) 1752 (C=O); 1HNMR (CDCl3, 500 MHz): δ (ppm) 4.61 (2H, s, -CH2), 7.11-7.17 (2H, m, 2-F-benzyl H3 & H4), 7.34-7.51 (3H, m, H8 & 2-F-benzyl H5 & H6), 7.60 (1H, t, J=7.5 Hz, H6), 7.74 (1H, t, J= 7.2 Hz, H7), 7.83 (2H, d, J= 8.5 Hz, phenyl H2 & H6), 8.14-8.16 (3H, m, H5 & phenyl H3 & H5), 16.31 (1H, s, -OH); LC-MS (ESI) m/z: 475.0 (M+1, 100). Anal. Calcd. for C24H15FN4O4S; C, 60.76; H, 3.19; N, 11.81. Found: C, 60.90; H, 3.03; N, 11.97.
3-((4-(5-(4-Fluorobenzylthio)-1, 3, 4-oxadiazol-2-yl) phenyl)diazenyl)-4-hydroxy-2H-chromen-2-one (11h)
Yield 64%; Orange powder; mp 246.6-251.6 °C; IR (KBr disk): υ (cm-1) 1763 (C=O); 1HNMR (CDCl3, 500 MHz): δ (ppm) 4.56 (2H, s, -CH2), 7.07 (2H, t, J= 8.5 Hz, 4-F-benzyl H3 & H5), 7.35-7.41 (2H, m, H6 & H8), 7.49-7.51 (2H, dd, J= 8.2 Hz, 5, 4-F-benzyl H2 & H6), 7.75 (1H, t, J= 7.5 Hz, H7), 7.83 (2H, d, J= 8.9 Hz, phenyl H2 & H6), 8.14-8.15 (3H, m, H8 & phenyl H3 & H5), 16.31 (1H, s, -OH); LC-MS (ESI) m/z: 475.0 (M+1, 100). Anal. Calcd. for C24H15FN4O4S; C, 60.76; H, 3.19; N, 11.81. Found: C, 61.01; H, 3.37; N, 11.69.
| No. | Log P | MW | HBA | HBD | Violations | nROTB | TPSA | ABS% | Volume | Log S |
|---|
| 11a | 3.32 | 380.38 | 8 | 1 | 0 | 4 | 114.09 | 69.64 | 306.47 | -6.300 |
| 11b | 3.82 | 394.41 | 8 | 1 | 0 | 5 | 114.09 | 69.64 | 323.27 | -6.532 |
| 11c | 4.27 | 408.44 | 8 | 1 | 0 | 6 | 114.09 | 69.64 | 340.07 | -6.802 |
| 11d | 4.81 | 456.48 | 8 | 1 | 0 | 6 | 114.09 | 69.64 | 378.12 | -7.881 |
| 11e | 5.41 | 490.93 | 8 | 1 | 1 | 6 | 114.09 | 69.64 | 391.65 | -8.617 |
| 11f | 5.41 | 490.93 | 8 | 1 | 1 | 6 | 114.09 | 69.64 | 391.65 | -8.617 |
| 11g | 4.91 | 474.47 | 8 | 1 | 0 | 6 | 114.09 | 69.64 | 383.05 | -8.195 |
| 11h | 4.91 | 474.47 | 8 | 1 | 0 | 6 | 114.09 | 69.64 | 383.05 | -8.195 |
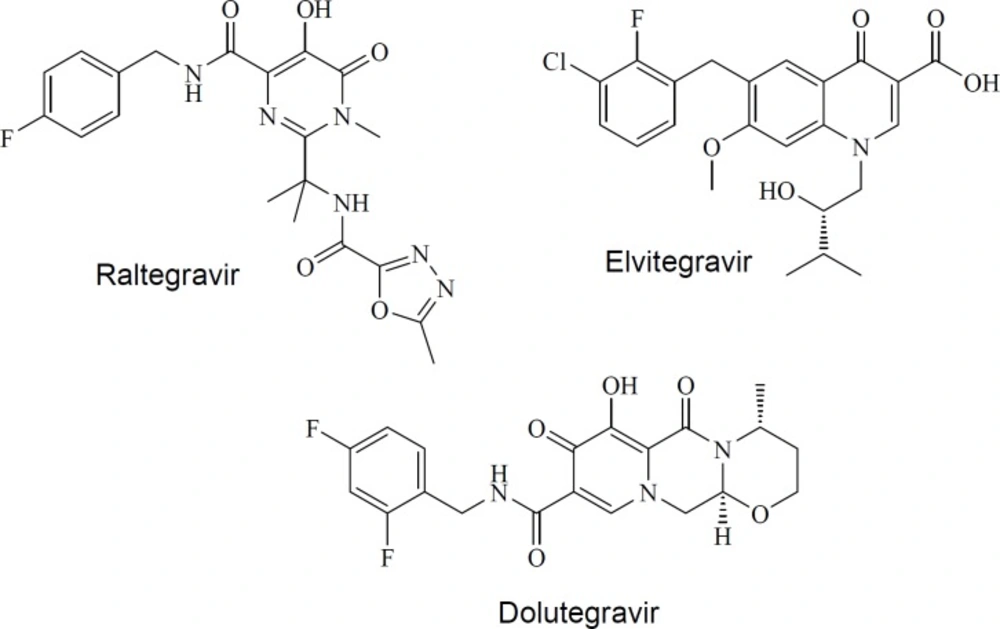
| RAL | -0.69 | 444.42 | 11 | 3 | 0 | 6 | 152.25 | 56.47 | 375.58 | -1.978 |
| ELV | 3.34 | 447.89 | 6 | 2 | 0 | 7 | 88.77 | 78.37 | 383.21 | -5.655 |
| DTG | 0.12 | 419.38 | 8 | 2 | 0 | 3 | 100.87 | 74.20 | 345.56 | -2.579 |
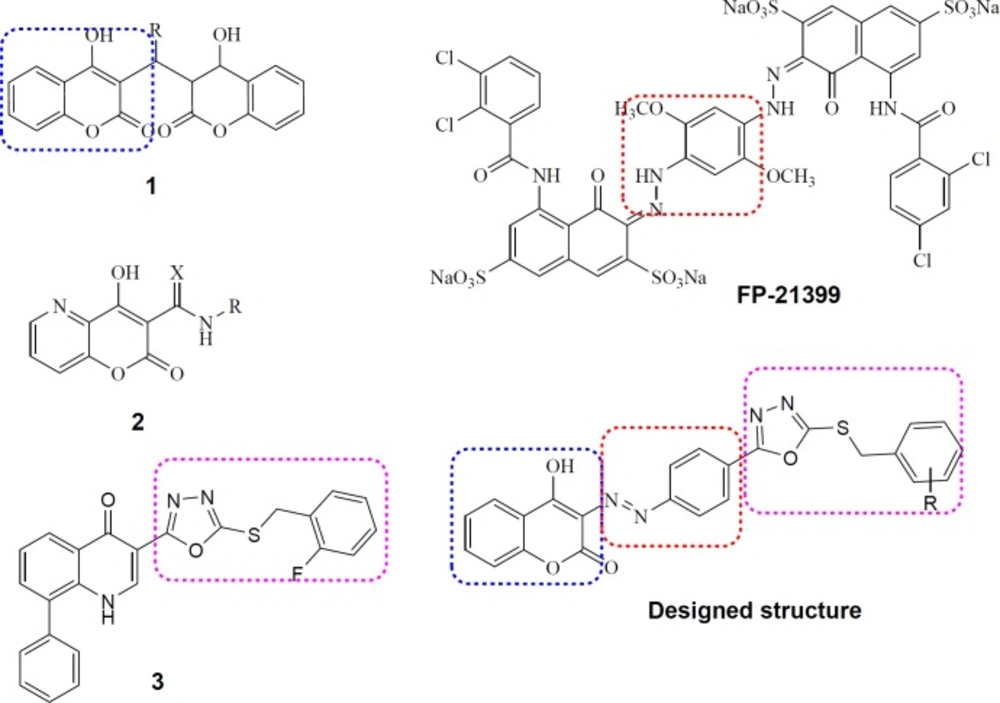
Lead anti-HIV compounds (1, 2, 3 and FP-21399) and our designed structure
Reagents and conditions: (a) ethanol, sulfuric acid, reflux, 24h (b) ethanol, NH2-NH2.OH (6 eq.), reflux, 96h (c) ethanol, CS2, KOH, reflux, 4h (d) methanol, 10% aq. NaOH, substituted alkyl/aryl halides, r.t (e) 1. 6M hydrochloric acid 2. 10% sodium nitrite, 0-5 °C (f) 10% aq. Na2CO3, stir, 0-5 °C
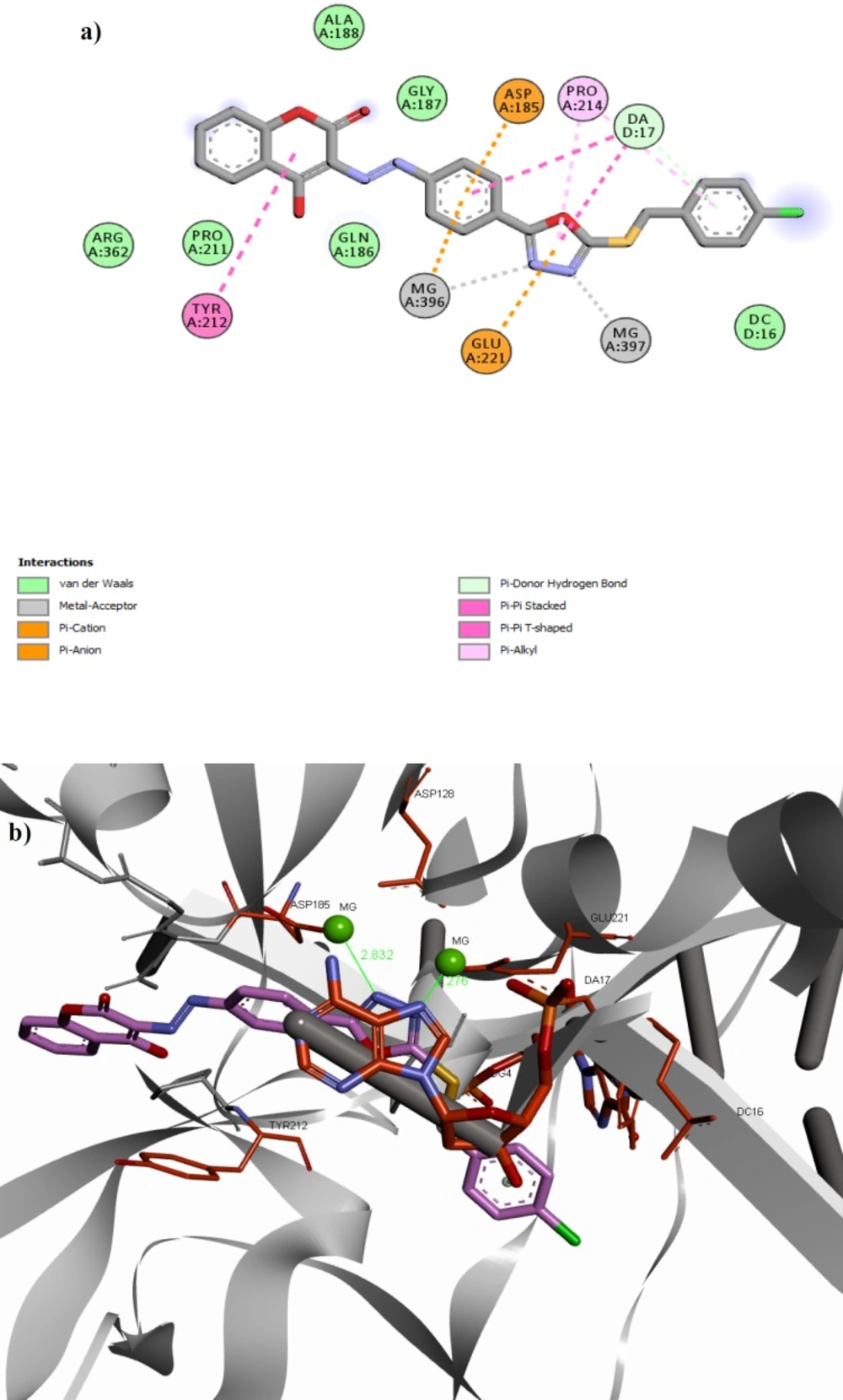
(a) 2D alignment of best docked conformer of compound 11f and (b) 3D alignment of best docked conformer of compound11f (shown in pink) in the PFV IN active site
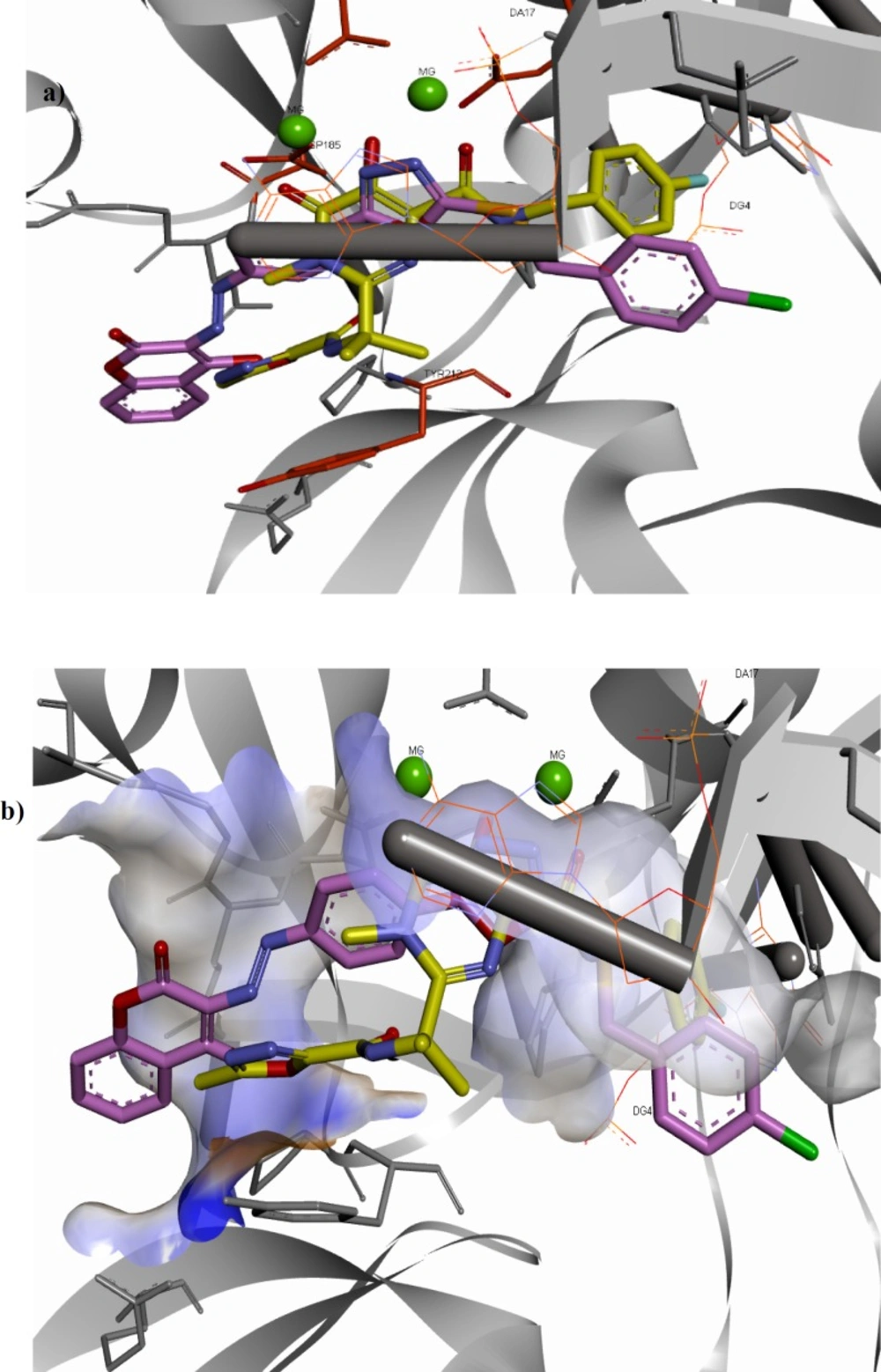
(a) Superimposition of compound 11f (shown in pink) on raltegravir (shown in yellow) in the PFV IN active site. (b) Interaction of compound 11f (shown in pink) and raltegravir (shown in yellow) with the surface of PFV IN active site designated with aromatic property
Molecular modeling studies
Molecular modeling was performed using the Autodock Vina (
28). According to the literature, X-ray crystallographic structure of prototype foamy virus (PFV) IN (PDB: 3OYA) in complex with DNA, two Mg
2+ cations and raltegravir has many similarities with secondary structure of HIV-IN (RMSD 1.04 Å). Thus, it could be considered as a good model for the development of INSTIs (
29). The protein and ligands were prepared in Autodock tools 1.5.6 from MGL Tools package (
30). The co-crystallized ligand and water molecules were extracted, Kollman charges were added, nonpolar hydrogens were merged and AutoDock4 atom type assigned to the protein structure. The ligand was created and minimized using HyperChem 8.0 (
31). The active site was defined as a Grid box around the crystallographic ligand raltegravir in 20×20×20 dimensions. All molecules were docked in the active site and the bioactive conformations were generated using Autodock Vina.
In vitro anti-HIV and cytotoxicity assays
Anti-HIV-1 activity of synthesized compounds was evaluated in single cycle replication assay which was evaluated in our laboratory and reported previously (
32-
35). In this assay, the single-cycle replicable HIV NL4-3 virions (200 ng p24) were inoculated concurrently with compounds in different concentrations to the Hela cells. The inhibition rate (%) of p24 expression was measured by capture ELISA (Biomerieux, France) 72 hours after inoculation. Percentage inhibition of p24 expression in treated culture was calculated as inhibition rate of p24 (%). The XTT (sodium3-[1(phenylaminocarbonyl)-3,4-tetrazolium]-bis(4-methoxy-6-nitro)benzene sulfonic acid) proliferation assay was conducted to evaluate the cellular toxicity according to kit instructions (
36,
37). Cytotoxicity test was performed exactly after the p24 assay.