All chemicals used in this study were of analytical grade purity and used as received. 2-chlorotrityl chloride resin (100-200 mesh) and N-α-fluorenylmethyloxy carbonyl (Fmoc) amino acid derivatives were purchased from NovaBiochem (Laufelfinegen, Switzerland). The necessary solvents with peptide grade synthesis were purchased from Sigma-Aldrich Company (St. Louis, MO, USA). The microbial culture media, trypticase soy agar (TSA), trypticase soy broth (TSB) and heart infusion broth (BHI) were commercially available from Merck (Germany). The cell culture medium RPMI 1640 supplemented with 10% fetal bovine serum (FBS), amino acids, vitamins and antibiotics from Gibco (Germany) was used for culturing cell line HT-29.
Preparation of microcinJ25-peptide derivative
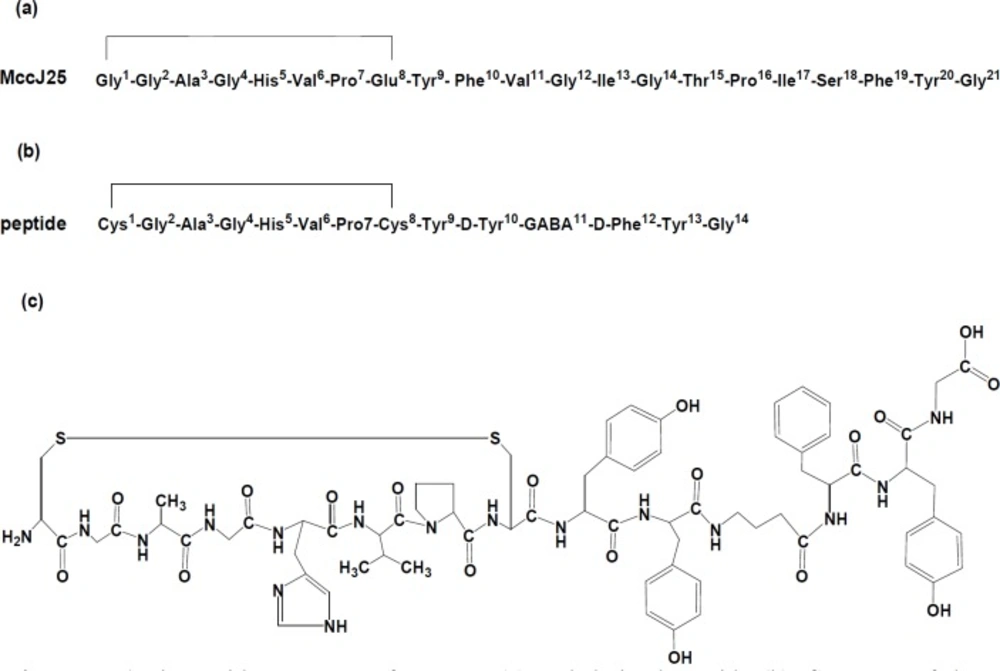
The protected peptide was constructed using standard Fmoc-based solid phase synthesis on 2-chlorotrityl [(2-Cl) Trt] resin with substitution, 1.4 mmol/g (
30). The amino acid derivatives with and without side chain protecting groups were respectively as: Fmoc-Cys(Acm)-OH, Fmoc-Gly-OH, Fmoc-Ala-OH, Fmoc-Gly-OH, Fmoc-His(trityl)-OH, Fmoc-Val-OH, Fmoc-Pro-OH, Fmoc-Cys(Acm)-OH, Fmoc-Tyr(tBu)-OH, Fmoc-D-Tyr(tBu)-OH, Fmoc-GABA-OH, Fmoc-D-Phe-OH, Fmoc-Tyr-(tBu)-OH, Fmoc- Gly-OH. Briefly, peptide bond formation between amino acids was made in the presence of 3 mole excess Fmoc-amino acid, 3 mole excess N-hydroxybenzotriazole (HOBt), 3 mole excess diisopropylcarbodiimide (DIC) and 9 mole excess diisopropylethylamine (DIPEA) in dimethylformamide (DMF). Completeness of amino acids coupling was monitored by the Kaiser test. The Fmoc protecting groups were removed by treatment with 20% piperidine solution (v/v) in DMF (10 mL). The synthesized peptide was cleaved from resin by the cleavage solution containing 20% trifluoroethanol (TFE), 0.5% trifluoroacetic acid (TFA), and 0.5% water (H2O) in dichloromethane (20 mL), subsequently incubated at room temperature for 30 min. Thereafter formation of disulfide bond between first and eighth cysteine amino acids was performed by iodine oxidation method. After dissolving Cys-Acm peptide (26 mmol) in 10% aqueous methanol (80 mL), the iodine solution (380 mg I
2 in 2 mL Methanol) was added. After 30 min vigorous stirring, quench of iodine was performed by addition of 0.5 M ascorbic acid (0.44 g, 5 mL). The mixture was evaporated under reduced pressure to dryness and redissolved in 40 mL DCM and 10 mL H
2O. Cyclic peptide was extracted in DCM phase and dried under reduced pressure. The side chain deprotection was performed by dissolving the peptide in a mixture of TFA (92.5%) /Thioanisole (2.5%)/ Triisopropylsilane (2.5%)/ H
2O (2.5%) for 30 min at 25 ˚C. After removing the organic solvents in vacuum, the crude product was precipitated in cold petroleum ether and Diisopropyl Ether (50:50).
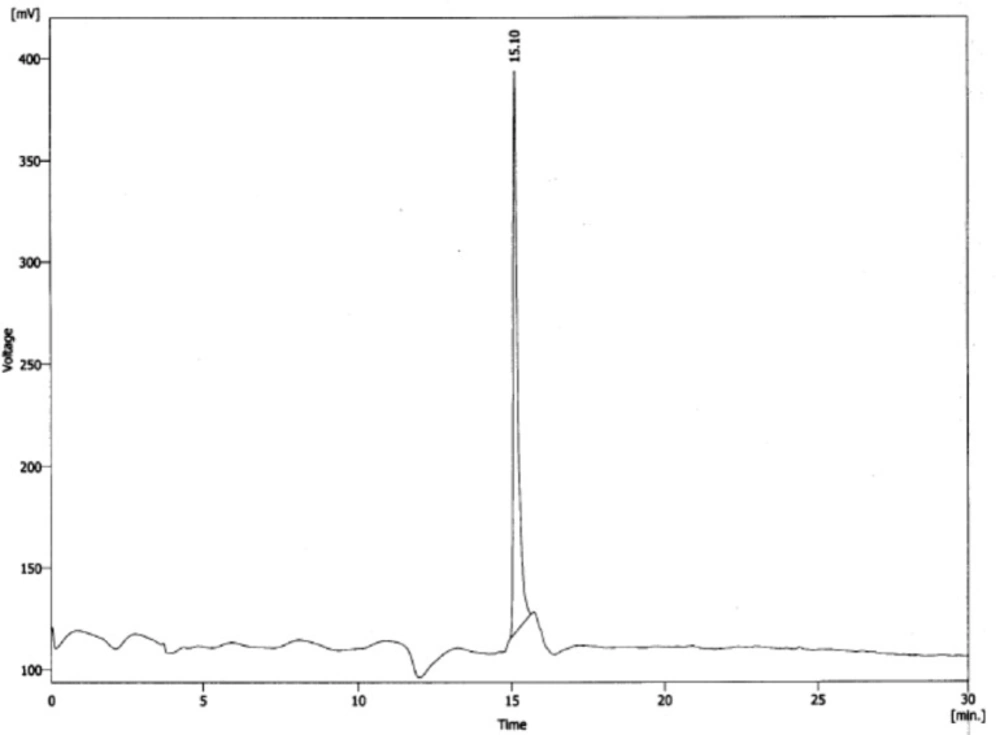
Purification of the peptide performed by a semi-preparative HPLC system (Sykam S7131, Eresing, Germany) using column VP Nucleosil 100-5 C18 (250×21 mm), flow: 15 mL/min with a variable wavelength UV detector operating at λ=214 nm. The mobile phase consisted of 0.1% trifluroacetic acid (TFA)/H2O (solvent A) and acetonitrile (solvent B) with gradient: 0 to 5 min, 70% A; 5 to 15 min, 70% to 30% A; 15 to 20 min, 30% A. Finally, the purified peptide was characterized by analytical HPLC system using a C18 column (CC 250/4.6 Reprosil-pur ODS-3.5 µm), flow: 1 mL/min; λ=280 nm (0.1 % trifluoroacetic acid/water (Solvent A) and acetonitrile (Solvent B) and the gradient: 0 to 5 min, 95% A, 5 to 25 min, 95% to 0% A, 25 to 27 min, 0% A, 27 to 30 min 0% to 95% A). The peptide was stored at -20 ˚C until use.
Structural determination of MccJ25 peptide derivative
For identification of peptide, LC-MS was performed by using Agilent G6410 triple Quadra pole mass spectrometer instrument in positive electrospray ionization (ESI) mode with Nebulizer: 15psi and LC condition as follows: Peptide solution (5 µL) was injected to LC- mass spectrometer using C18 column and mobile phase consisted of water/0.1%formic acid (solvent A) and methanol (solvent B). The gradient used a flow rate of 0.3 ml/min with a column temperature of 25 ˚C in A: B ratio of: 50:50. For further confirmation, FTIR spectrum of the product was recorded by a Brucker Vector 22 spectrometer instrument (Germany).
Antibacterial activity assay
Bacterial strains and growth conditions
All bacteria were obtained from a microbial laboratory collection (Iran University of Medical Sciences). All strains were maintained in 20% glycerol/Brain Heart Infusion broth (Merck, Germany) at -70 ˚C. Two Gram-positive bacteria (Staphylococcus aureus ATCC25923, Staphylococcus aureus PTCC1112) and four Gram- negative bacteria (Salmonella enterica PTCC1709, Salmonella typhimurium ATCC14028, Escherichia coli ATCC35218 and Escherichia coli ATCC8739) were used in this study. All selected bacterial strains were grown aerobically in Trypticase Soy Broth (TSB) (Merck, Germany) at 35-37 ˚C. Stock cultures of the bacterial strains were maintained on Trypticase Soy Agar (TSA) (Merck, Germany) slants and stored at 4 ˚C. The strains were sub-cultured in fresh media at every experiment interval.
Bacterial suspension standardization
A McFarland 0.5 standard was prepared by mixing 99.5 mL of 0.18 M sulfuric acid (1% v/v) with 0.5 mL of 0.048 M (1.175%) barium chloride and was dispensed into screw cap glass tubes. The tubes were tightly sealed and stored under dark conditions at room temperature. The standard tubes were agitated on a vortex mixer before use. The McFarland 0.5 standard provides turbidity (OD
630 = 0.08-0.1) comparable to that of a bacterial suspension containing approximately 1-2× 10
8 colony-forming units (CFU/mL) (
31).
Preparation of bacterial suspension
For preparation of bacterial suspension, 4-5 colonies of overnight culture of the test bacterial strain were transferred to 5 mL of a liquid growth medium (TSB) and the tubes (in a similar sized glass tube to the standard) were incubated at 35-37 ˚C for 2-3 h. After incubation, the turbidity of suspension was adjusted to 0.5 McFarland tubes by using UV/visible spectrophotometer (Biowave П, Biochrom,China).
Determination of Minimal Inhibitory Concentration (MIC)
The broth micro-dilution method using sterile 96-well microplate was performed for evaluation of minimal inhibitory concentration (MIC) of the peptide derivative as recommended by Clinical and Laboratory Standards Institute (CLSI, formerly NCCLS) (
31). A stock solution of peptide (422 µM) was prepared in ultra-pure deionized water and then the different concentrations of the peptide on the basis of serial two fold dilutions ranging from 0.97-250 µM were obtained from stock solution. The microplate wells were loaded with 100 µL of bacterial suspension (1.5-2× 10
8 CFU/mL in TSB medium, OD
630= 0.09-0.1) and followed by the addition of 10 µl peptide solution. Each peptide concentration was tested in triplicate on microplate. Wells containing bacterial cells in medium without peptide (positive control) and wells containing TSB medium and peptide without bacterial cells (negative control) were included on the same plate. The blank wells were loaded with TSB medium. The optical density of the plate was measured at 630 nm by using a microplate reader (BioTek, USA) at two steps as follows: after loading the wells and after 24 h incubation at 37 ˚C, respectively. MIC values were determined as the lowest peptide concentration that caused 100% inhibition of bacterial growth based on optical density of each well. Commercial antibiotic gentamicin at concentration (0.97-500 µM) was used as a standard for evaluation of MIC. In addition, the inhibitory effect of the highest concentration of peptide (250 µM) on bacterial growth (bacterial suspension containing 1.5-2× 10
8 CFU/mL) was evaluated for 24 h incubation at 35-37 ˚C.
Cell culture
HT-29 tumor cell line used in this study was purchased from Pasture Institute of Iran. The cells were maintained in RPMI 1640 (Gibco, Germany) supplemented with 10% (v/v) FBS, L-glutamine (2 mM), penicillin G (100U/mL) and streptomycin (100 µg/mL). The cells were cultured at 37 °C in a humidified incubator with 5% CO2.
Cytotoxicity assay
In-vitro cytotoxicity of the peptide derivative on cells was assayed using the MTT [3-(4, 5-dimethylthiazoyl-2-yl) 2, 5 diphenyltetrazolium bromide] method (
25). HT-29 cells were plated in a sterile 96 wells polystyrene microplate (10
4- 10
5 cells/well). Microplate was incubated at 37 °C in 5% CO
2 atmosphere for 48 h. After removing the medium, each well loaded with 100 μL of fresh medium and 20 µL of different concentrations of the peptide in sterile ultra-pure deionized water in range from 0.1 to 250 µM. The wells without peptide were used as a negative control. Following 24 h incubation under the above mentioned conditions, the medium removed by vacuum and then filter-sterilized MTT (100 μL, 5 mg/mL DMSO) was added to each well and incubated for 3 h. The media was removed and the content of the wells for dissolving formazan crystal was mixed with 100 μL DMSO. Optical density was measured at 540 nm by using microplate reader and cell viability percentage was calculated by the absorbance ratio of cells treated with peptide to untreated cells dissolved in complete media. The experiment was performed in triplicate format for each peptide concentration under the same conditions.
Hemolysis assay
1 mL of fresh human blood from a healthy donor (age 48, male) with sodium citrate as anticoagulant was used for measuring the peptide derivative hemolytic activity. The prepared blood was centrifuged at 500 g for 5 min at 5 °C for the separation of red blood cells (erythrocytes). The separated red blood cells were washed three times with sodium phosphate saline solution (PBS, pH 7.3) and then the blood sample was diluted to a final concentration 5% (v/v) with PBS. Red blood cells suspension (100 µL) was added to 10 µL of different concentrations of the peptide derivative ranging from 0.1- 250 µM in centrifuge tubes and incubated at 37 °C for 1 h. Each peptide concentration was tested in triplicate format on microplate. 1%Triton X-100 solution and PBS solution were used as positive and negative control respectively. After incubation, the tubes were centrifuged at 1000 g for 5 min. 100 µL of the supernatant were added to the wells of 96- well microplate. The absorbance of the samples was measured at 540 nm by using microplate reader. The percentage of hemolysis was calculated by following formula as described by Hemmami
et al. (
26). Hemolysis (%) = [(mean Absorbance value of treated sample × mean Absorbance value of negative control) / mean Absorbance value of positive control)] × 100.
Statistical analysis
Statistical evaluation was carried out by using the SPSS 11.5 version for windows. All results were expressed as mean values ± SD (n = 3) for three independent experiments. Statistical significance was defined as p < 0.05.

![Mass spectrum analysis of the prepared peptide derived from MccJ25 in positive electrospray ionization (ESI) mode. Peaks at m/z 1519.9 and 1560.8 correspond to molecular ions [M+H] + and [M+CH3CN+H] + respectively](https://brieflands.com/journals/ijpr/articles/126167/figures/ijpr-18-1264-g003-preview.webp)