Introduction
Experimental
Results
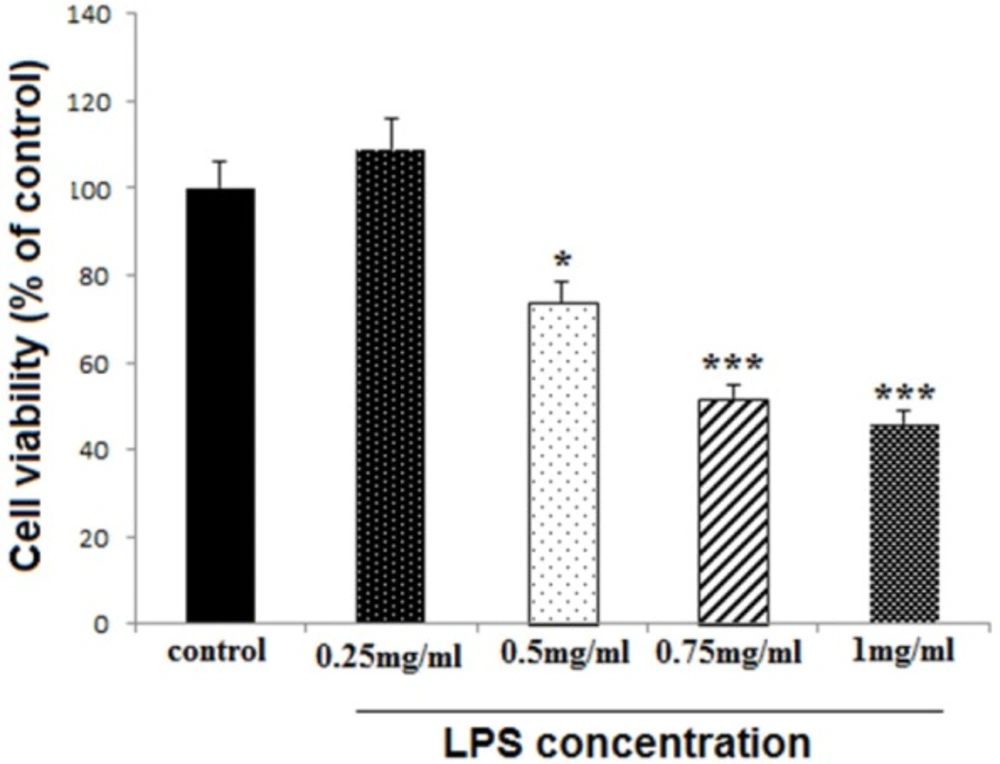
Effect of high dose LPS on cell viability in neural like cells. Cells (1 × 104/well) were treated with LPS at 0.25, 0.5, 0.75 and 1 mg/mL concentrations for finding toxic dose, and then, cell viability was evaluated MTT assay. LPS reduced cell viability at 0.5, 0.75 and 1 mg/mL concentrations, while the lower concentration did not change cell viability. Data are shown as mean ± SEM. Treatments were repeated 3 times and there were 4 wells in each repeat. *P < 0.05, ***P < 0.001 vs. Ctrl
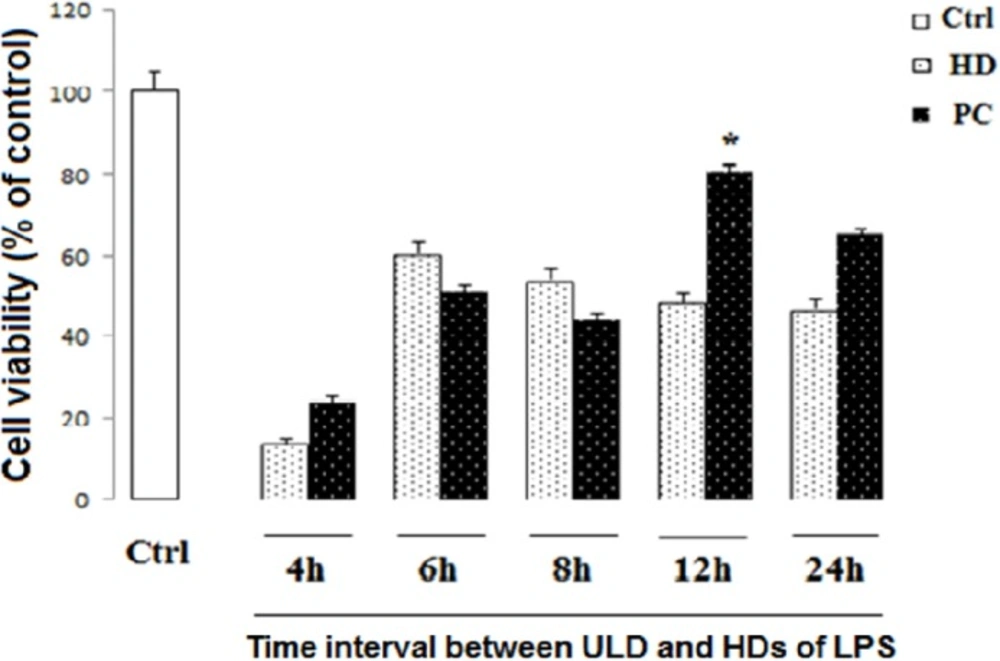
Effect of PC and time of incubation on cell viability in neural like cells. Cells (1 × 104/well) were treated with HD LPS and ultra-low dose (ULD) LPS for PC induction for 4, 6, 8, 12, and 24 h, and then, cell viability was measured using MTT test. Cell viability reduced following HD LPS. PC increased cell viability following 12 h interval between HD and ULD, while other intervals did not change cell viability. Data are shown as mean ± SEM. Treatments were repeated 3 times and there were 4 wells in each repeat
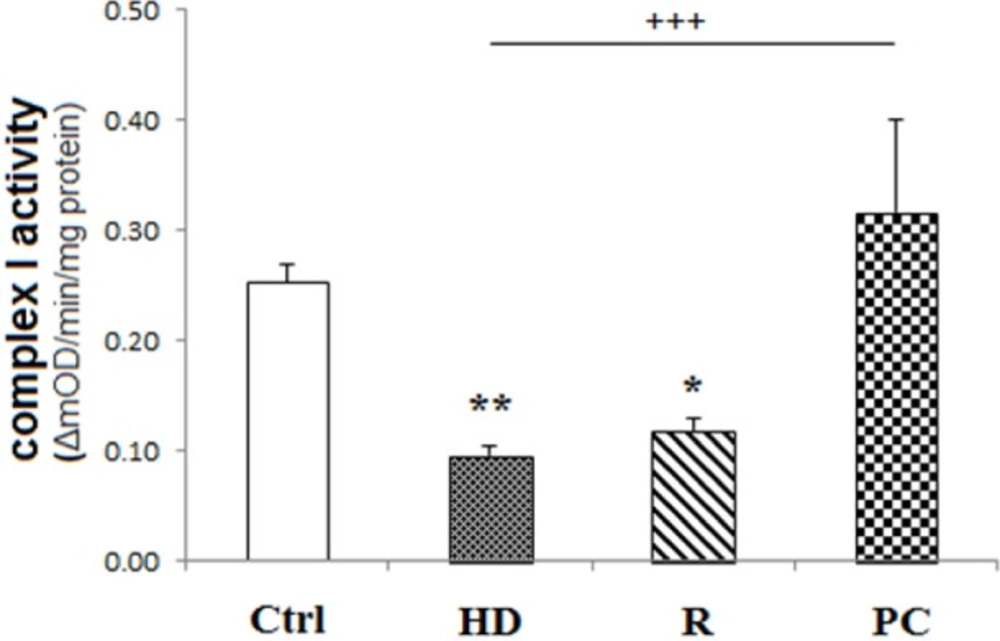
High dose LPS reduced the activity of complex I. HD LPS reduced complex I activity while PC preserved this activity. Data are shown as mean ± SEM. Treatments were repeated 3 times. *P < 0.05, **P < 0.01 vs. Ctrl, +++P < 0.001 vs. HD
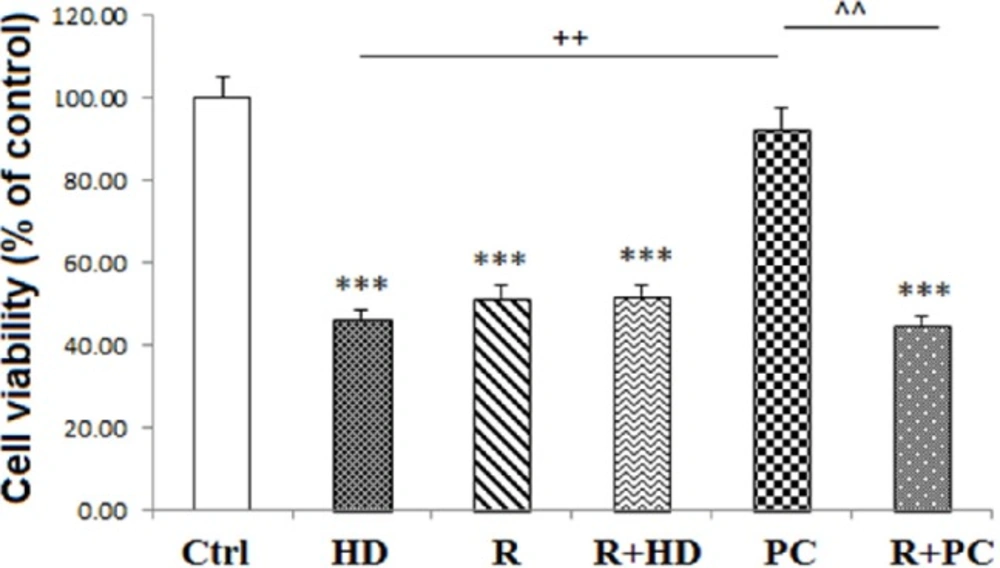
Effect of inhibition of complex I and PC on neural like cells measured using MTT test. Inhibition of complex I attenuated the protection effect of PC on cell viability. Data are shown as mean ± SEM. Treatments were repeated 3 times and there were 4 wells in each repeat. ***P < 0.001 vs. Ctrl, ++P < 0.01 vs. HD, ^^P < 0.01 vs. PC. Ctrl: Control; HD: high dose lipopolysaccharide (LPS); PC: preconditioning; R: Rotenone
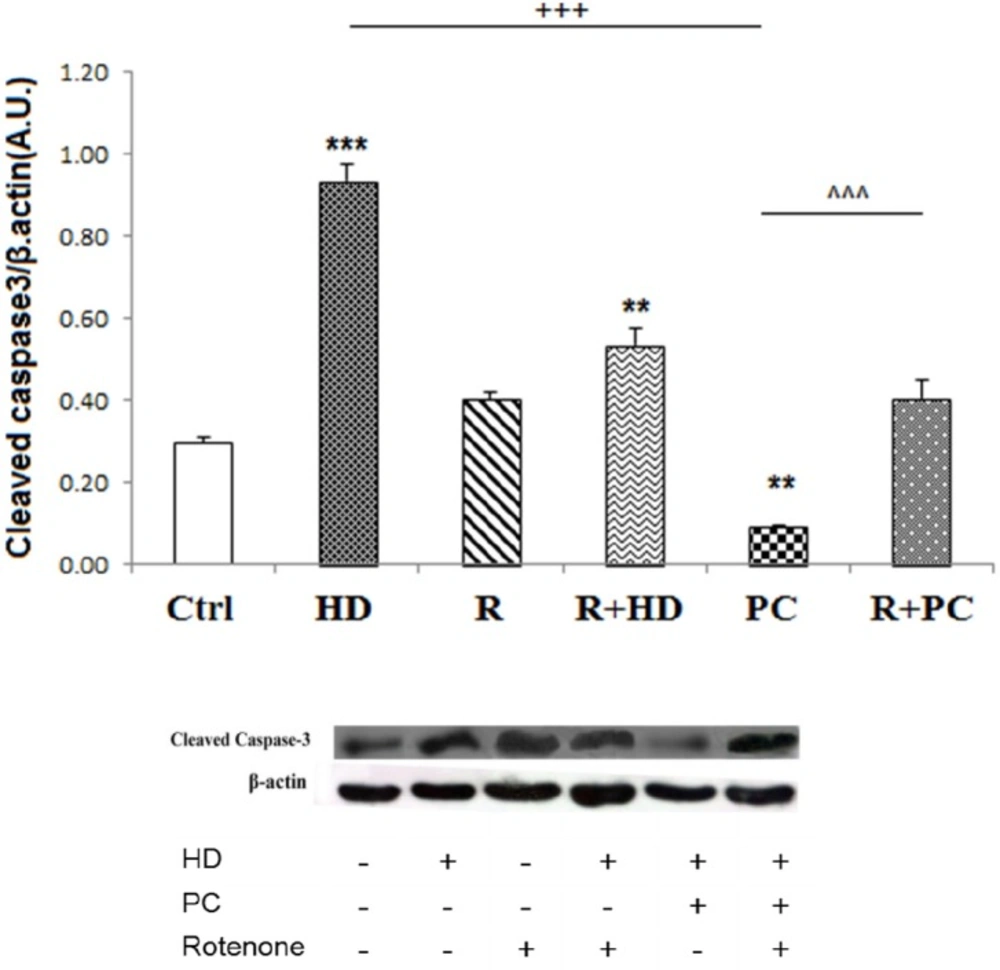
The role of complex I in protective effect of PC against caspase 3 cleavage induced by high dose LPS. Inhibition of complex I by rotenone increased caspase 3 cleavage. PC prevented the activation of apoptotic pathways in. response to high dose LPS. Data are shown as mean ± SEM. Treatments were repeated 3 times. **P < 0.01; ***P < 0.001 vs. Ctrl, +++P < 0.001 vs. HD, ^^^P < 0.001 vs. PC. Ctrl: control; HD: high dose lipopolysaccharides (LPS); PC: preconditioning; R: Rotenone