Brain dissection and cellculture
All experimental procedures were approved by the institutional Animal Care and ethical committee of Shahid Beheshti University of Medical Sciences.
Primary hippocampal cell cultures were prepared from 80 brains of 2-4 day-old (P2-P4) Wistar rats according to the method described by Brewer
et al. (
28). Twenty replicates were done (four replicates for each experimental group) and in each replicate four rat pups were used. Briefly, pups were decapitated after being anesthetized on ice, and hippocampi were dissected into ice-cold dissociation buffer containing calcium and magnesium free Hank’sbalanced salt solution (0.976%), sodium bicarbonate (0.035%, Sigma, UK) and pyruvate (1 mM), HEPES(10 mM) with a pH 7.4. Then, the tissue was gently triturated (15-18 times) through a fire-polished Pasteur pipette until it was dispersed into a homogenous suspension. The dispersed cells were centrifuged for 5 min at 1000 rpm, Neurons were then cultured on glass coverslips (16 mm in diameter) pre-coated with poly-L-lysine (0.01% poly-L-lysine ,Sigma UK) in a B27/neurobasal medium containing B27 (2%), neurobasal (96.75%), and L-glutamine (0.5 mM, Sigma, UK without antibiotic at a density 1-2 × 10
5 cell/cm
2. Antibiotics were not applied to the nutrient medium because they could affect the spontaneous activity (29). The cells were incubated at 37 °C under 5% CO
2 and fed twice weekly. The morphological changes and growth of the neurons were observed using an Olympus IX71 inverted microscope (Olympus, Japan). The electrophysiological properties of cultured hippocampal neurons were recorded at day 14
in-vitro (DIV-14) using whole-cell patch-clamp method, under current and voltage clamp conditions.
Whole-cell patchclamp of cultured hippocampal neurons
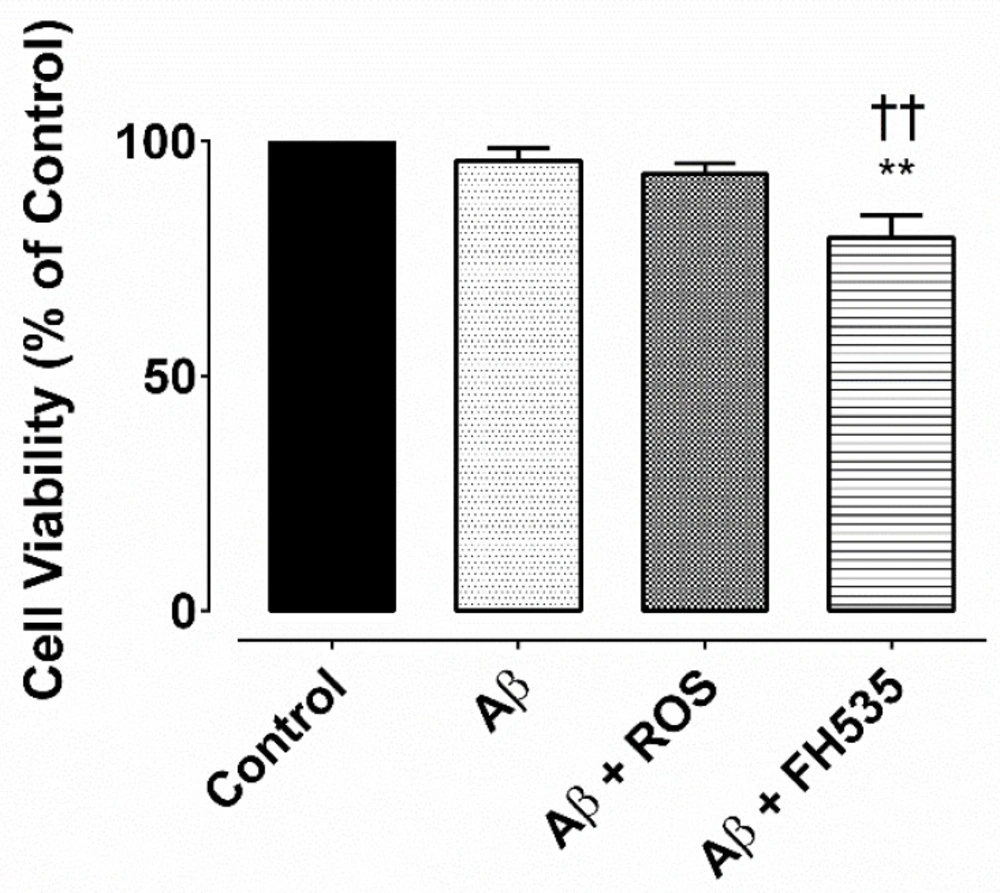
The cultured neurons were divided into four groups (n > 50 neurons in each group): control, Aβ alone-treated, Aβ+rosiglitazone (ROS) and Aβ+FH535 treated. Aβ
1-42 was prepared by dissolving the peptide in sterile PBS to a final concentration of 100 nM and incubated at 37 °C for 27 h. Rosiglitazone (30 µM) and FH535 (15 µM) were dissolved in DMSO. The final concentration of DMSO was 0.1% (v/v). At this concentration, it had no effect on neuronal activity (data not shown).After twelve days in culture, the neurons were exposed to either Aβ alone or in combination with rosiglitazone or FH535 for 24 h and then the whole-cell recordings were made as previously described (
29). Briefly, a coverslip with cultured pyramidal neurons was placed in a recording chamber mounted on an inverted microscope (Olympus IX71, Japan) equipped with an Olympus DP12 camera and continuously perfused with HEPES-based artificial cerebrospinal fluid containing (in mM) 140 NaCl, 2 CaCl
2, 1.4 KCl, 10 HEPES, 10 glucose, pH 7.3 (adjusted with NaOH) and osmolarity was 295-297 mOsml/L, bubbled with 100% O
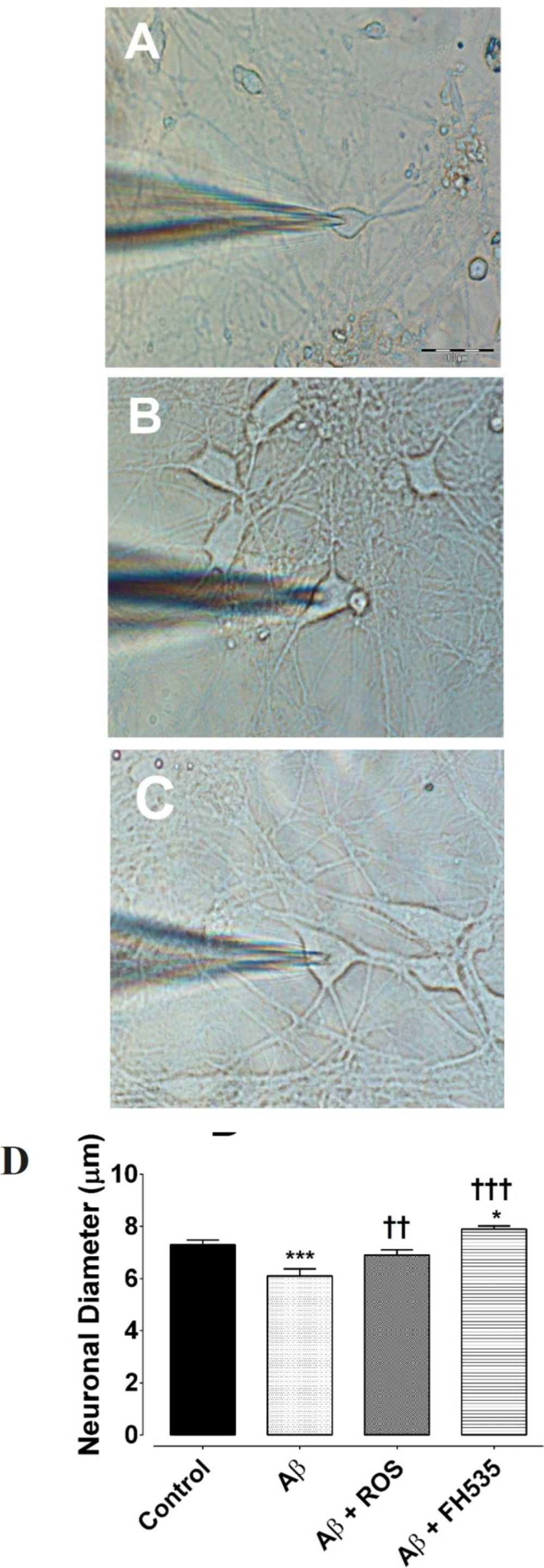
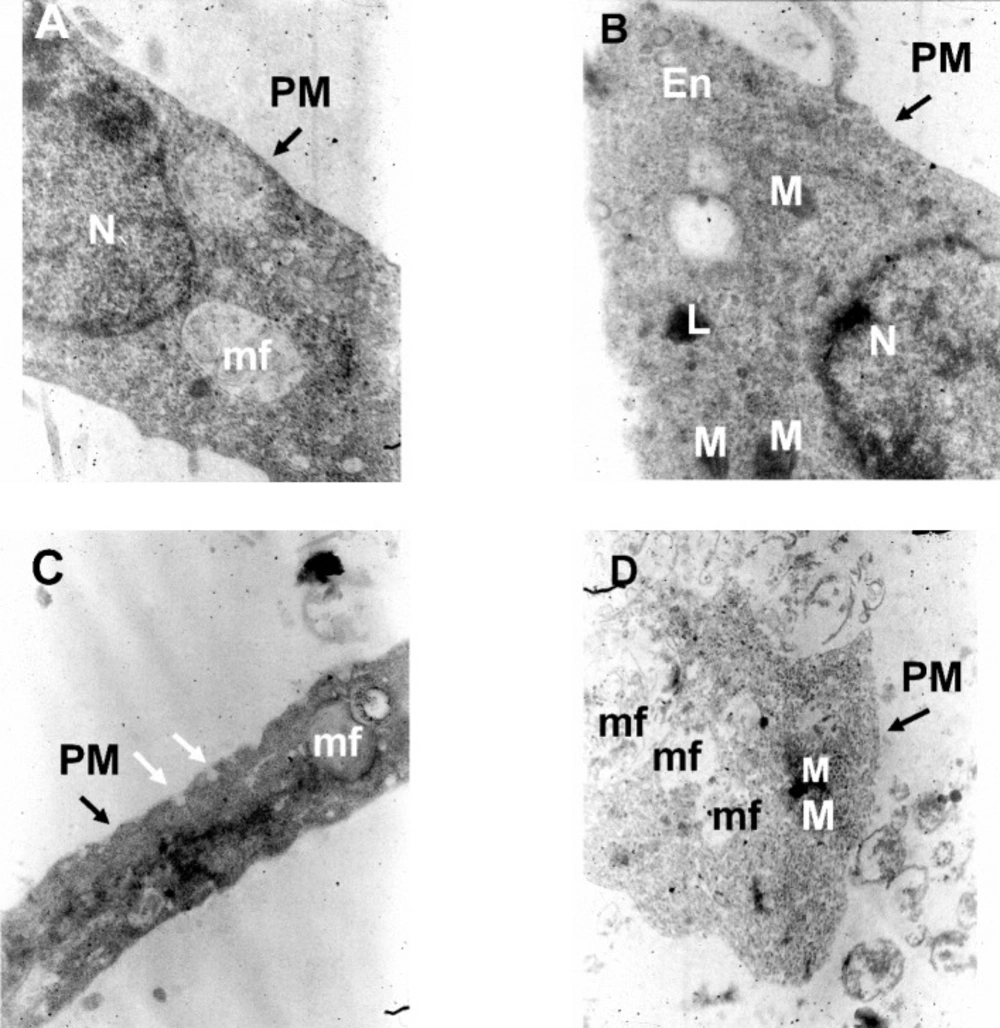
2, and continuously flowed throughout the experiments at 2 mL/min. Cultured hippocampal pyramidal neurons were identified by their morphological characteristics (
Figures 1A-1D). The micropipettes were pulled by a two-stage puller (PP-10, Narishige, Japan) from borosilicate glasses (1.5 O.D, 0.86 I.D.) with a tip resistance of 5-7 MΩ when filled with internal solution containing (in mM) 145 KCl, 3 NaCl, 0.5 CaCl
2, 10 HEPES, 2 Na
2 ATP, and 0.4 Na
2 GTP. The pH of the internal solution was set to 7.3 by KOH, and the osmolarity was adjusted to 280-290 mOsm.
All recordings were made at room temperature (22-25 °C) using a Multiclamp 700B amplifier (Axon Instruments, Foster City, CA). Data were filtered at 5 kHz and sampled at 10 kHz using a Digidata 1440 analog-to-digital convertor and pClamp 10 software (Axon Instruments, Foster City, CA) and stored on a personal computer for offline analysis. Recordings were discarded when the series resistance was over 20 MΩ.
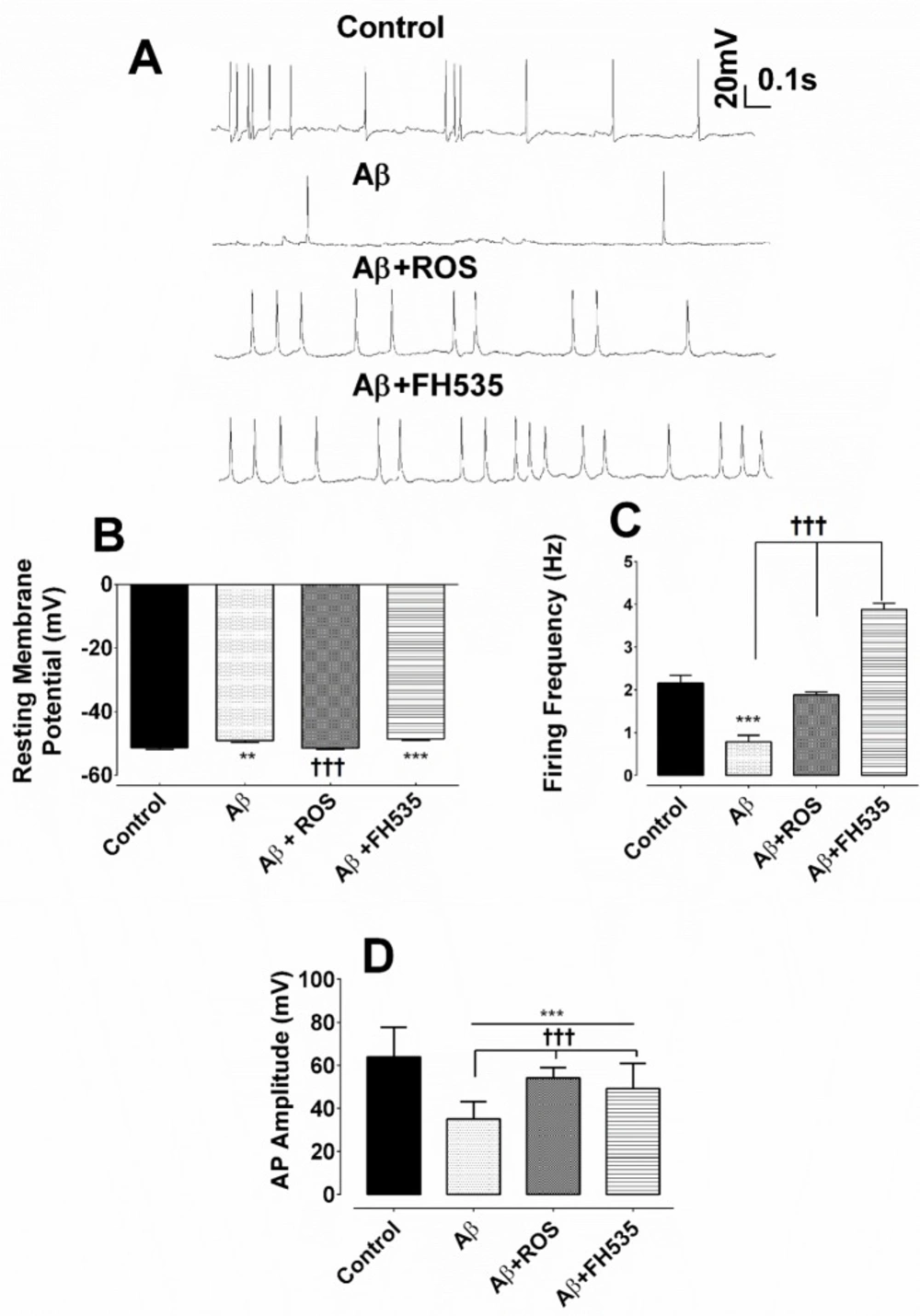
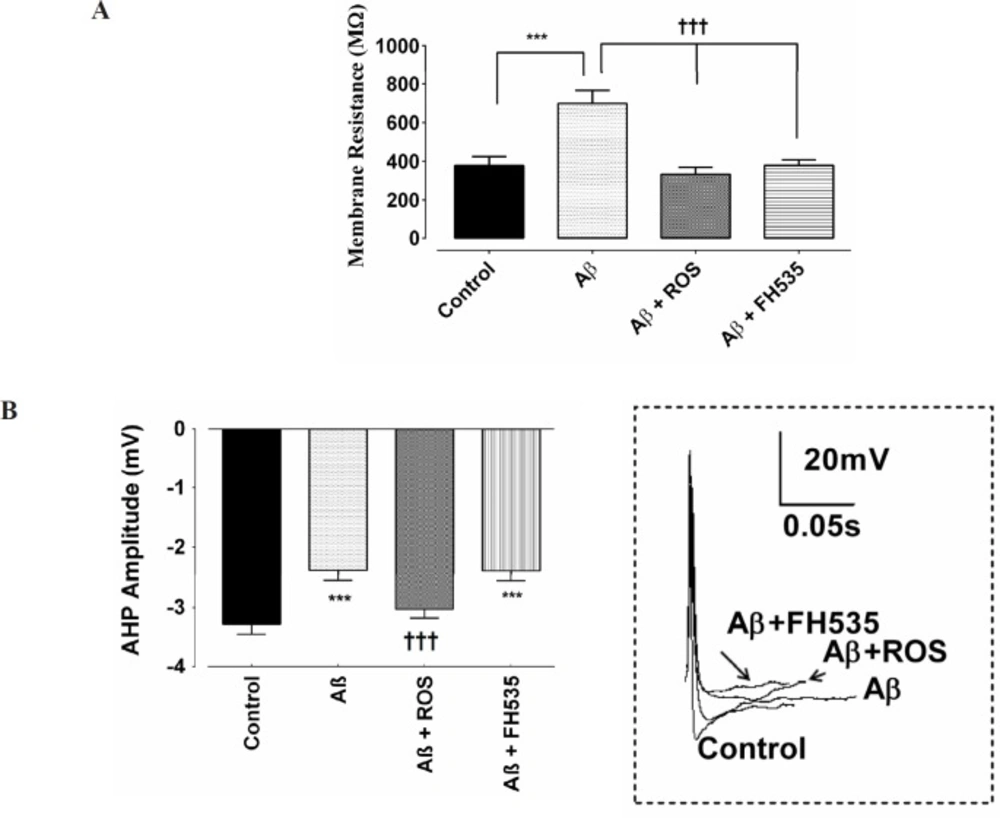
The resting membrane potential (RMP), the membrane resistance (Rin), the firing frequency, and the after hyperpolarization (AHP) were measured under current clamp condition. The AHP amplitude was measured from the RMP to peak of AHP. Mean firing frequency was calculated as the number of action potentials over the 1 sec period. Rin was determined as the slope of the linear current-voltage relationship.
Voltage-clamp experiments
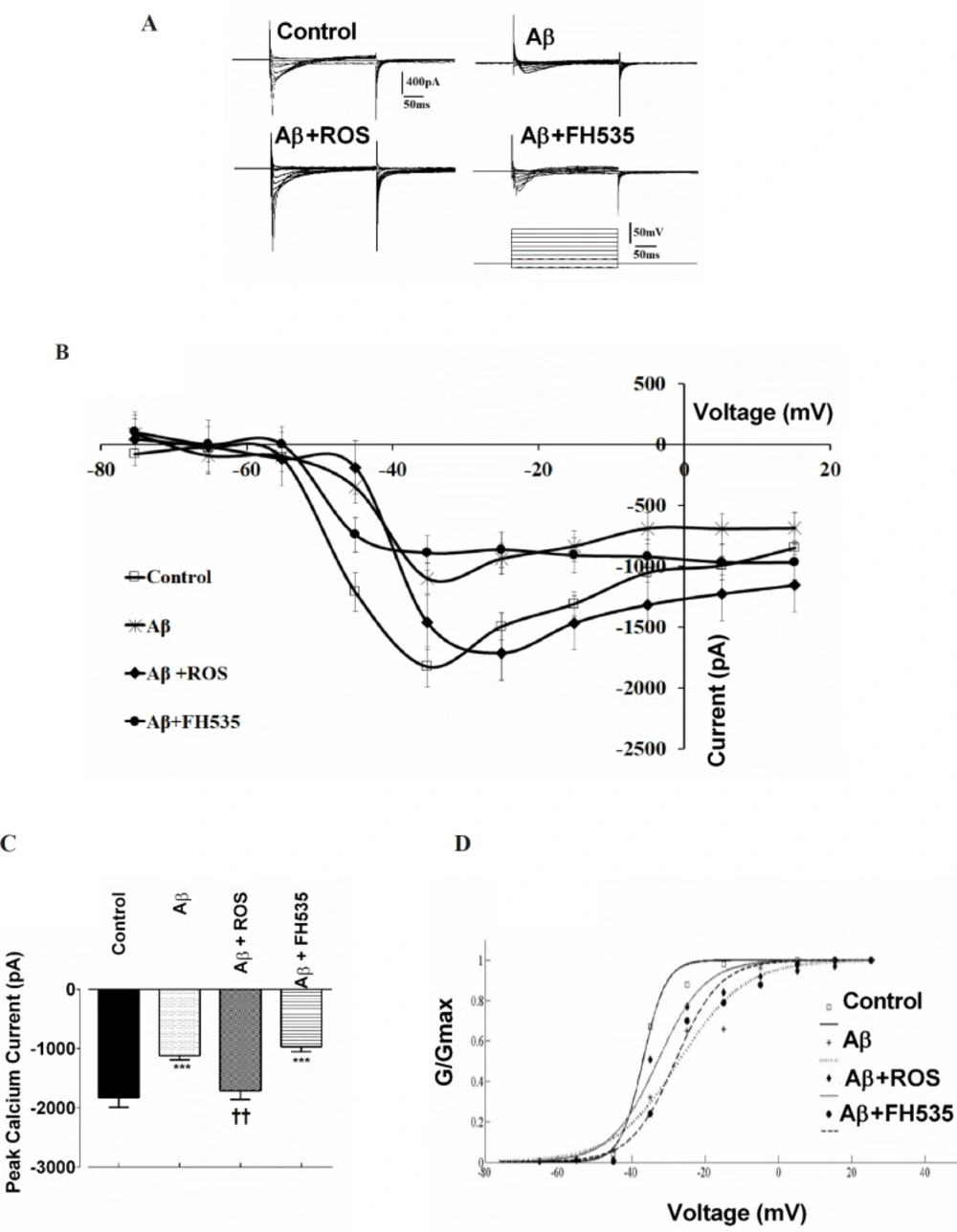
Inward Ca2+ currents were recorded with a pipette containing (in mM): 10 CsCl, 100 Cs2 MeSO4, 0.5 CaCl2, 10 HEPES, 0.5 EGTA, 2 Na2 ATP, 0.3 Na2 GTP, 5 QX-314, and, 20 TEA; pH 7.4 adjusted with CsOH, 295-297 mOsm/L. The extracellular ACSF solution for recording Ca2+ currents consisted of (in mM): 126 NaCl, 1.4 KCl, 2.5 CaCl2 , 10 Hepes, 10 Glucose , 3 Pyruvic acid and 2 mM 4-AP and 10 mM TEA were added to block outward K+ channel currents. The pH was adjusted with NaOH to 7.4 and osmalarity was 280-290 mOsmol/L. Ca2+ currents were elicited by voltage steps from -75 mV to +15 mV in 10 mV increments from a holding potential of -65 mV for 2500 msec. Furthermore, to assess the changes in the voltage sensitivity of Ca2+ channel current activation, the half maximal activation (Vhalf) was determined by fitting individual conductance- voltage (G-V) relationship with a Boltzmann function using MATLAB (The MathWorks):
Where the Gmax is the mean value of maximal conductance, Vhalf is the voltage for half-maximal activation of channels and k is the slope factor. The channel conductance was calculated according to the equation:
G = ICa2+ /(Vm-Vrev)
where G is the conductance, ICa2+ is the Ca2+ tail current, Vm is the holding voltage, and Vrev is the reversal potential of ICa2+. Vrev was determined to be about 100 mV by linear extrapolation to the peak of the tail current from clamping at voltages between -75 mV to 25 mV.
Morphometric and electron-microscopic methods
In order to characterize changes in cultured neuronal dimension the longest axis of the soma was measured. Eighty cultured pyramidal neurons were randomly chosen to measure their soma size (20 cell in each experimental groups). The ultrastructure of the cultured neurons was also evaluated at day 14 under electron microscope, as described by Bahrami et al. (30). Briefly, after two weeks, the cultured pyramidal neurons on plastic cover slips in different experimental conditions were fixed in 2.5% glutaraldehyde for 1 h with osmium tetroxide post-fixation for 30 min at 37 oC then, rinsed with 3.6% NaCl and water. Thereafter, the cells were dehydration in graded ethanol and embedded in LR-white resin and after polymerization at 60 oC for 3 days, the grids were made. Next, the sections were stained with uranyl acetate and lead citrate and observed with a Zeiss electron microscope.
Assessment of cell viability
Cell viability was performed as previously described using Cell Titre 96 Aqueous One Solution (MTS) Cell Proliferation Assay Kit (Promega Ltd.), which measures mitochondrial activity (29). Briefly, cultured neurons on coverslips either in control condition or following treatment with Aβ, Aβ +ROS, or Aβ + FH535 were washed with phosphate buffer saline (PBS). Then,100 µL of the serum-free culture medium containing 20 µL of mixture ofMTS[3-(4,5-dimethylthiazol-2yl)-5-(3-carboxymethoxyphenyl)-2(4-sulfophenyl)- 2H-tetrazolium] and PMS [phenazine methosulfate as the electron coupling reagent] was added into each well of the 96-well assay plates and re-incubated for 2 h at 37 ºC. The absorbance of the sample was recorded at 490 nm using a Wallac microplate reader. The MTS reduction values were expressed as percentage of the control (untreated cells). The percentage of viable cultured cells was then plotted as percentage of control.
Statistical analysis
The data were checked for normal distribution using a Kolmogorov–Smirnov test and then analysedusing one way ANOVA followed by Tukey post-hoc test.
The values were presented as mean ± SEM and significant difference between experimental groups was at p < 0.05.