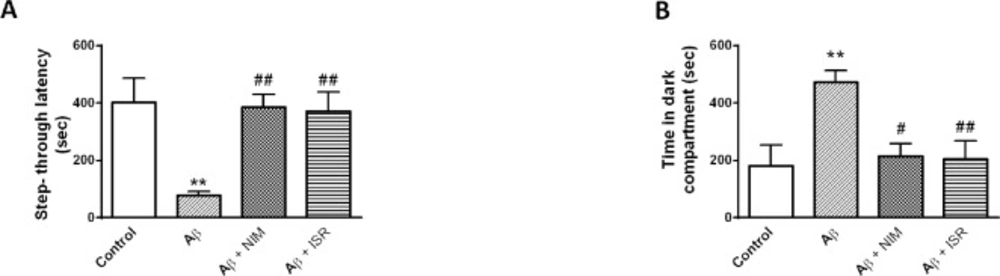Adult male Wistar rats (Pasteur Institute; Tehran, Iran) weighing between 220 ± 20 g at the time of surgery were used. The animals were housed four per standard cage, in a colony room with a 12/12 h light-dark cycle (7:00–19:00 lights on) at 22 ± 1 °C. Commercial rodent pellets and tap water were available ad libitum. They were allowed to adapt to the laboratory conditions for at least one week before surgery. The rats were handled about 5 m each day prior to behavioral test. Ten animals were used in each group of the experiments. A total number of 40 animals were used in the experiments. All procedures were carried out in accordance with the institutional guidelines for animal care and use.
Drugs and Surgery
The drugs used in the study were ketamine and xylazine (Alfasan Chemical Co, Woerden, Holland) for animal anesthesia, Aβ1-42 (Tocris, UK), isradipine, and nimodipine (Tocris, UK). Aβ was dissolved in 0.1 M phosphate-buffered saline (PBS; pH 7.4) and then kept at -70 °C until use. One week before the beginning of the behavioral experiments, the animals were anesthetized and Aβ or vehicle (DMSO) were bilaterally microinjected into the entorhinal cortex under the stereotaxic surgery (AP: -5.05, L: ± 6.6 and DV: -8.2) according to the Paxinos and Watson Atlas (
30). Injections were done over 2 min using a 30-gauge blunt-tipped needle attached to a 5-μL Hamilton syringe, and to avoid fluid back flow the needle remained in place for an additional 1 min. After entorhinal injection, a cannula (8 mm, 23 gauge) was implanted 1 mm above the right ventricle (AP: -0.96, L: 1.8, DV: -3.4) and then secured to the skull using dental cement. Over one week, the rats were treated daily with i.c.v. microinjection of nimodipine, isradipine (both 30 μg/μL, Tocris, UK), or vehicle (DMSO, Sigma-Aldrich,
USA).
The animals were divided into four groups (n = 10 each group): (a) Control group; given single microinjection of PBS (2 μL) into the entorhinal cortex, and daily DMSO (2 μL) microinjection into the right ventricle over one week. (b) DMSO-Aβ group; treated by Aβ (1 µg/2 μL) in the entorhinal cortex, and treated by daily microinjection of DMSO (2 μL) into the right ventricle for one week. (c) Aβ + ISR group; received Aβ (1 µg/2 μL) in the entorhinal cortex and daily treated by isradipine (30 µg/2 μL) for one week. (d) Aβ + NIM; received single microinjection of Aβ into the entorhinal cortex and daily microinjection of nimodipine (30 µg/2 µL) into the lateral ventricle.
Behavioral testing
Passive avoidance learning and memory
The Shuttle box consisted of two compartments, light and dark compartment (20 cm × 20 cm × 30 cm) which were divided by a guillotine door (7 cm × 9 cm). The floor composes of stainless steel grids (2.5 mm in diameter) with one-centimeter intervals and connected to an insulated stimulator. In order to habituate to the apparatus, each animal was put in the light chamber and 10 sec later the guillotine door was opened to allow the rat to enter the dark chamber. When the animal entered the dark compartment, the door was closed, and the rat was returned back to its home cage. After 30 min, the rat was placed in the light chamber and 10 sec later the door was opened and when the animal entered the dark compartment, the door was closed and a foot shock (50 Hz, 1.5 mA for 1.5 sec duration) was applied. After 10 sec, the rat was taken and returned to its cage. Two min later, the rats were allowed to repeat the task and the training was completed when they avoided entering the dark compartment for 120 sec. After 24 h, the rats were again placed in the light compartment and after 10 sec the door was opened, and the step-through latency and the time spent in the dark compartment were recorded for 10 min.
Novel object recognition test
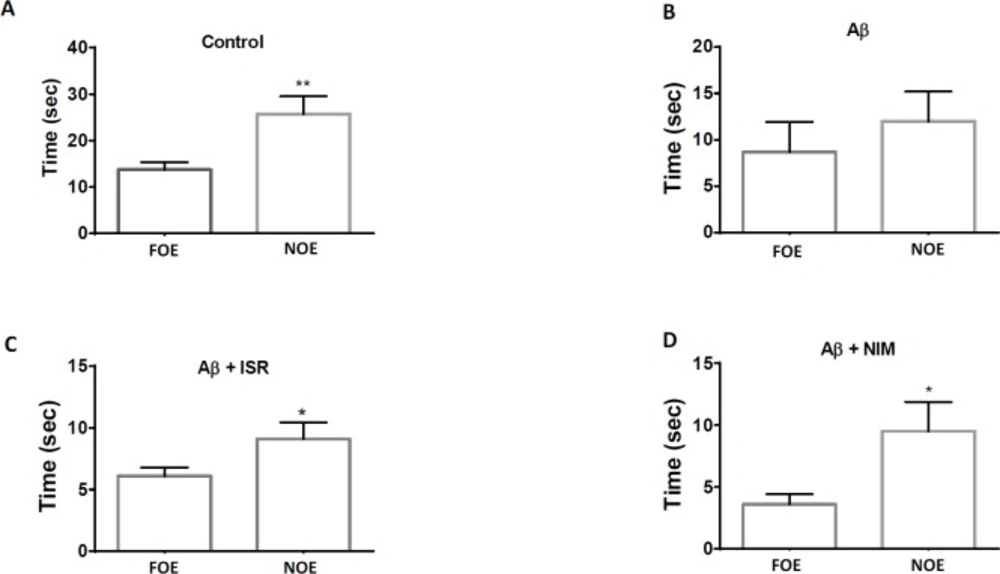
The Novel Object Recognition (NOR) test took place in a wooden open field box (40 × 40 × 60 cm). This test consists of the habituation phase, the familiarization phase, and the test phase. On the first day, the rats were habituated to the apparatus in which they were placed in the empty open field for 5 min. Twenty-four h later, the rats were allowed to explore two identical objects over 5 min. Twenty-four h after the training session, one of the object was replaced with a new object with distinctive shape and the rats were allowed to explore the open field for 5 min in the presence of two objects; the familiar object exploration (FOE) and a novel object exploration (NOE). Between trials, the objects were washed with 10% ethanol solution. The object exploration was measured using two stop watches to record the time spent exploring the objects during the experimental sessions.
Brain collection
After completion of the experimental sessions, each animal was decapitated and the right and left hippocampi were immediately removed and washed in the PBS. Then, the hippocampi were deep-frozen and stored at -80 °C until use. For BiP and CHOP protein level measurement, the animals were decapitated and their brains were horizontally sliced (500 µm) by vibroslicer (Campden Instruments, UK). Then, the dentate gyrus tissues were separated under loop microscope and homogenized in the lysis buffer containing a protease inhibitor cocktail and stored at -80 °C until use.
Western Blot Analysis
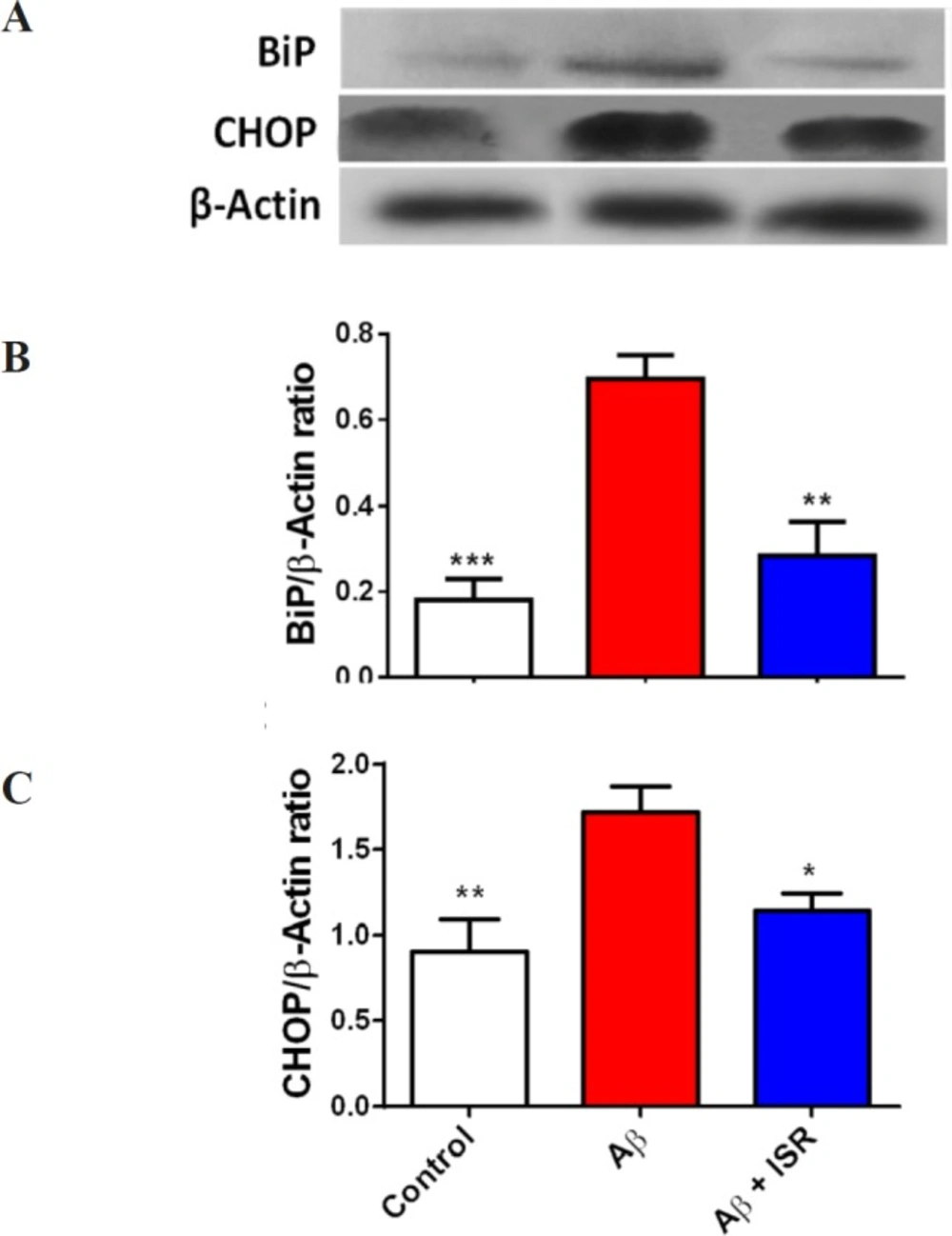
Protein concentrations of hippocampal and DG lysates were quantified using Bradford method to guarantee equal loading for assaying by electrophoresis and GSH assay (
31). They were lysed in lysis buffer containing complete protease inhibitor cocktail (Roche, Germany). For western blotting, 50 µg of DG total protein was electrophoresed in 12% SDS-PAGE gels, transferred to PVDF membranes and probed with anti-CHOP, anti BiP (1/1000 dilution, Cell Signaling Technology, Beverly, MA, USA), and anti β-actin (1/1000 dilution, Abcam, Cambridge, UK) as internal control. Electrochemiluminescence (ECL) reagents (Amersham Bioscience, USA) were used to detect immunoreactive polypeptides and then the results were quantified using densitometric scan of the films. Data analysis was done using ImageJ software (National Institute of Health).
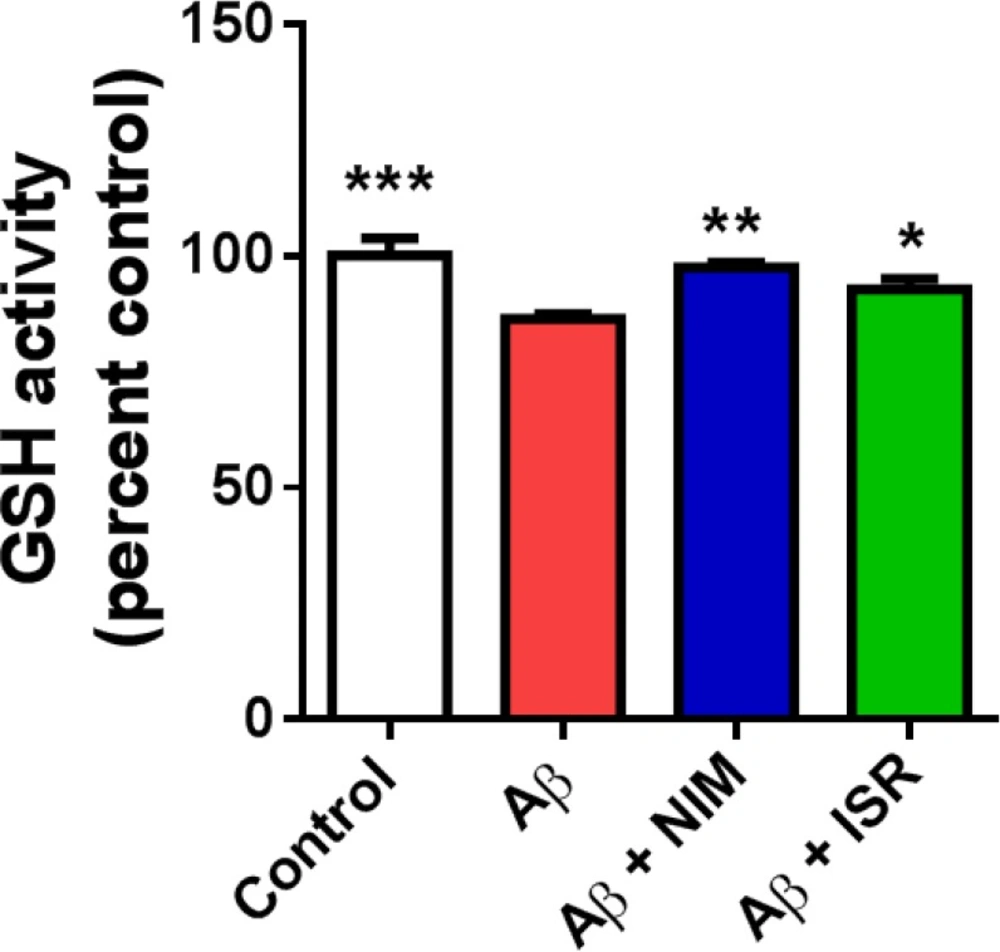
Determination of GSH activity
Glutathione reduction was measured using the Ellman’s methods (
32). Briefly, 100 μL of Ellman′s reagent (DTNB, 0.1 M) was mixed with 200 μL of hippocampus lysate and 0.25 mL sodium phosphate buffer (0.1 M). The amount of glutathione was measured by spectrophotometer method at 412 nm and the results were expressed as GSH/μg protein.
PDI assay
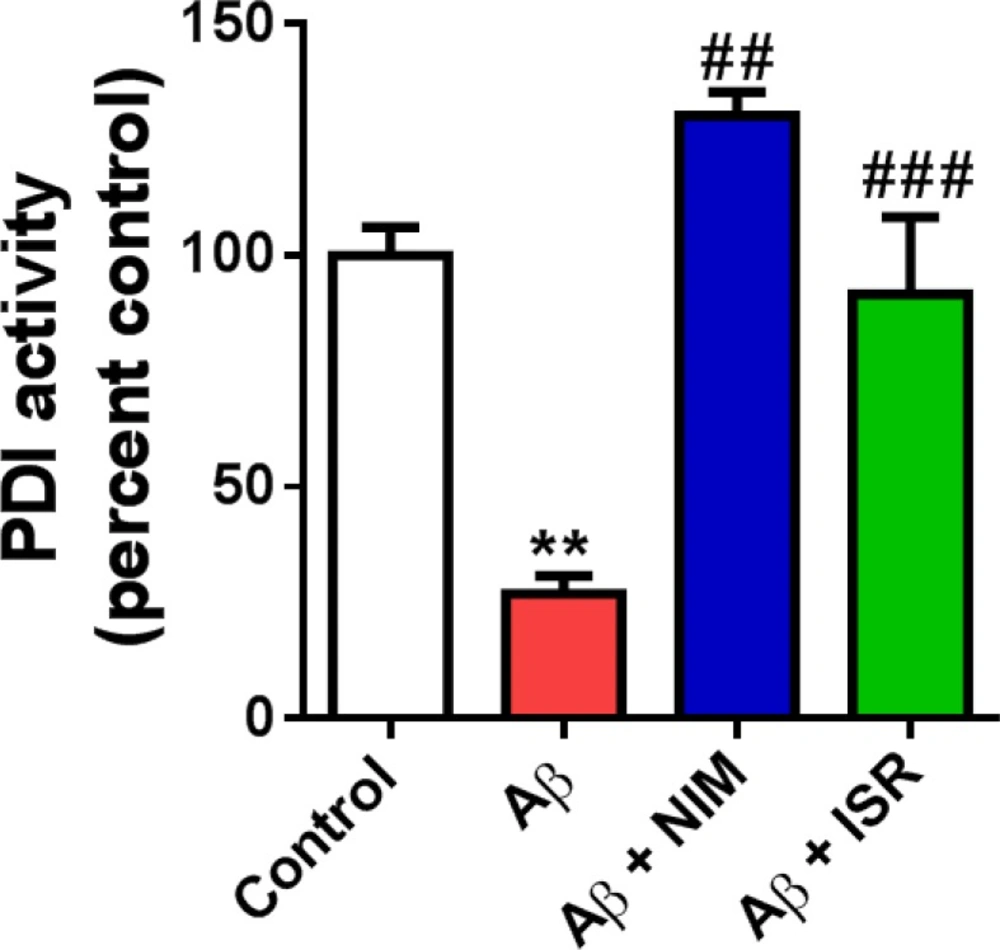
PDI activity was determined by measuring reduction of insulin in the presence of dithiothreitol (DTT) as a reducing agent. The cocktail contained 100 mM potassium buffer, 0.2 mM EDTA (pH 7.0), 0.15 mM insulin, and 1 mM DTT. Protein sample was added to solution and the polymerization of reduced insulin chains was traced at 650 nm (
33).
Data Analysis
The data were expressed as mean ± standard error of the mean and processed by GraphPad Prism 5.0. Results were statistically evaluated by One-way analysis of variance (ANOVA). Where F value was significant, post-hoc analysis (Tukey test) was performed. Object recognition test was analyzed using unpaired t-test. Differences with p < 0.05 between the groups were considered statistically significant.