Materials
Erlotinib-HCl (ELT) was purchased from Arasto Pharmaceutical Chemicals Inc (Tehran, IRI) miglyol were obtained from Gattefosse (St-Priest, France). Soybean phosphatidylcholine (PC) was achieved from Lipoid GmbH (Ludwigshafen, Germany). Dimethyl sulfoxide (DMSO, 99.9%),3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT), 4’,6-diamidino-2-phenylindole (DAPI), fetal bovine serum (FBS), RPMI-1640 medium (R5886), phosphate buffered saline (PBS 1X), trypsin (0.25% EDTA solution), poloxamer 407, tween 80Ò, cholesterol and ethanol 99% were supplied from Sigma Aldrich Co. (St. Louis, MO). The annexin V/PI apoptosis detection kit was purchased from Exbio Co. (Exbio, Czech Republic).
NLC preparation
NLC dispersions occurred by the hot homogenization technique. Firstly, ELT was dissolved in 200 m L DMSO. Then, the mixture was added to the melted solid and liquid lipid (Precirol and miglyol, respectively), and then homogenized under 10K rpm speed for 1 minute. Following that, the aqueous surfactant solution (poloxamer 407 with same temperature as melted lipids mixture) was gradually added to the lipid phase less than 23K rpm homogenization (Silent crusher M, Heidolph, Nuremberg, Germany) for 20 min. Then, the hot nano emollition was left in a stagnant place to chill (
21).
Liposome preparation
A thin layer film was used to produce liposome through hydration-sonication method. Soybean lecithin and cholesterol ratios (90:10, 80:20, 70:30, 60:40, 50:50 and 40:60 w/w) were mixed in 10 mL absolute ethanol as a solvent. Then, 5 mg of ELT was added to the lipid solution and mixed until complete dissolution. Then, the lipid/drug solution was poured into the round balloon. To provide the thin layer, the solvent was removed from the mixture by a rotary evaporator at 40
oC. The obtained film was hydrated by 10 mL of 1X PBS solution at 65
oC for 8 min. To reduce the size of the provided film, the formulation was exposed to a probing sonication (Vibra Cell-Sonics & Material, 130 W, 20 kHz, USA) at 80% sonication strength under ice bath for 10 min (10 cycles of 1 min sonication and 1 min rest for allowing the samples to cool down) (
22).
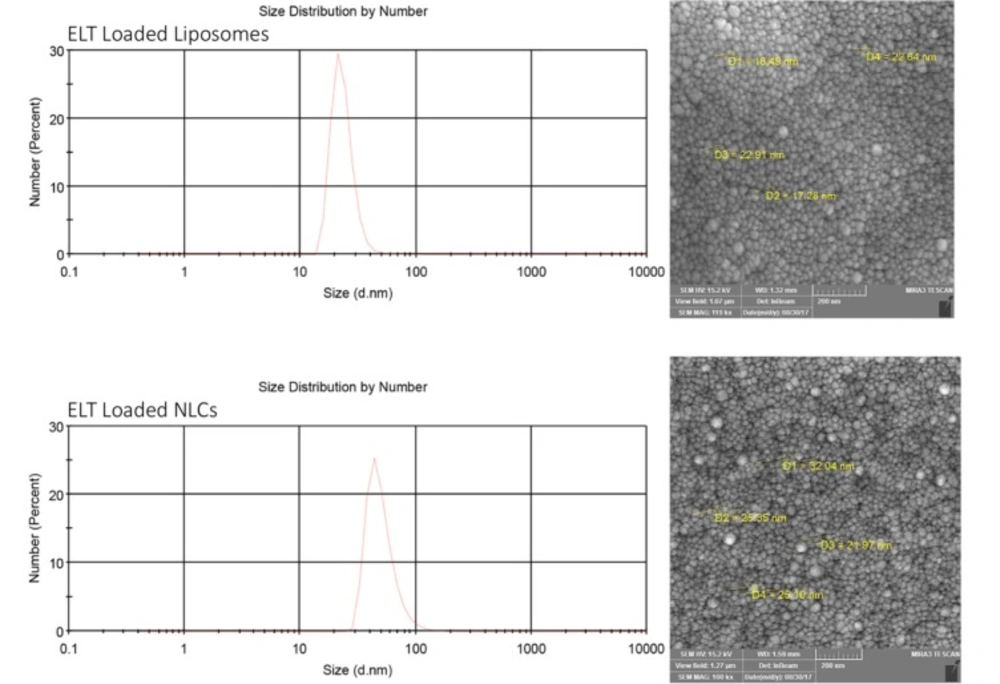
Size and zeta potential (ZP) analyses
The size and ZP of the nano liposomes and NLCs were measured by a dynamic light scattering (DLS) method using Malvern zeta sizer (Malvern Instruments Ltd., Malvern, UK) at the room temperature. All samples were diluted 10 times with purified water. All measurements were done in triplicate.
Scanning electron microscope (SEM)
The shapes and morphology of the prepared formulations were monitored by scanning electron microscope (TEscan, VEGA II XMU, Czech Republic). Before scanning, the samples were coated with gold, using a direct current sputter technique (DST1, Nanostructured coating co, Iran).
Encapsulation efficiency and drug loading
The EE parameter was determined by measurement of the concentration unentrapped ELT in NLCs and liposomes. The Amicon® Ultra-4 100 k – a 30 kDa molecular weight cut-off membrane (Millipore, Billerica, MA) was employed to this purpose. Briefly, 2 mL of each samples (ELT liposome and NLCs) were placed into the Amicon filter and centrifuged under 4K rpm for 10 min. The concentration of the unentrapped (unloaded) drug was determined by a validated UV–vis spectrophotometric method (Ultrospec 2000 UV/Visible, Parmacia Biotech Instruments Ltd, Cambridge, England) at 343 nm. The rate of DL and EE was calculated using the following equations (Equations 1 and 2), respectively:
where C
T, C
AP, W
DL, and W
NP represent total concentration of the added drug, concentration of drug in the aqueous phase, weight of the loaded drug into NPs, and solid mass weight of NPs, respectively (
23).
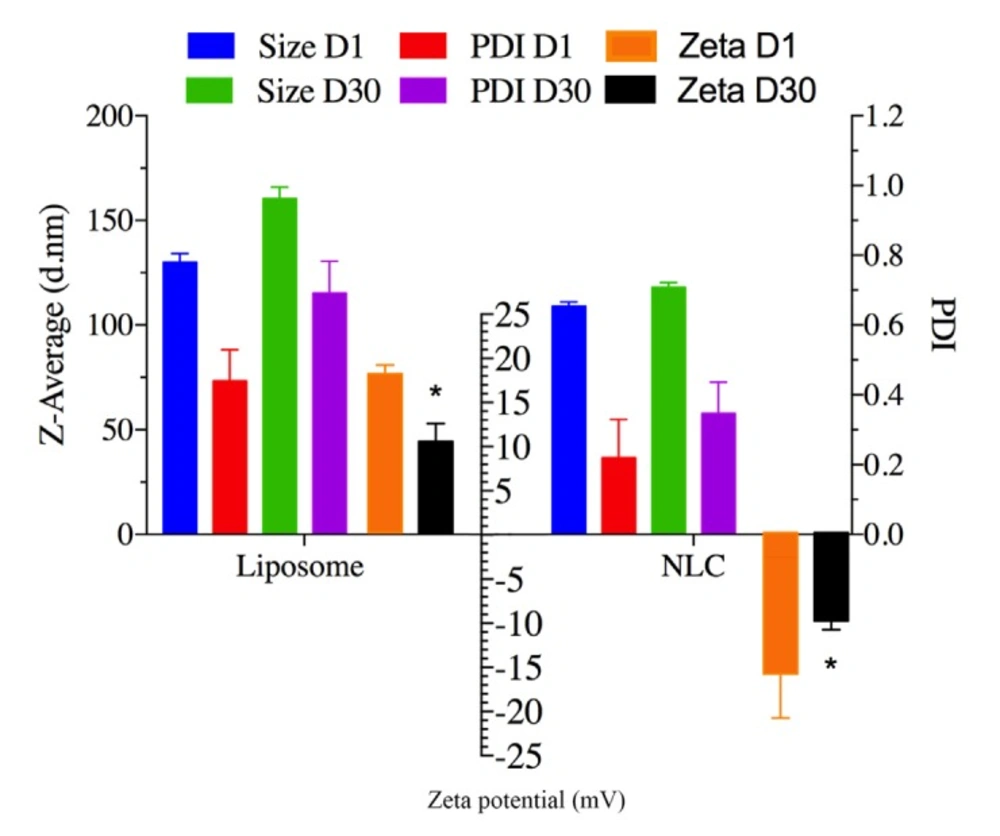
Stability study
Stability of ELT-liposome, ELT-NLCs was done by measuring changes in the particle size, ZP, and PDI. All of the samples were stored at refrigerator at 4-8 Co temperature for 30 days.
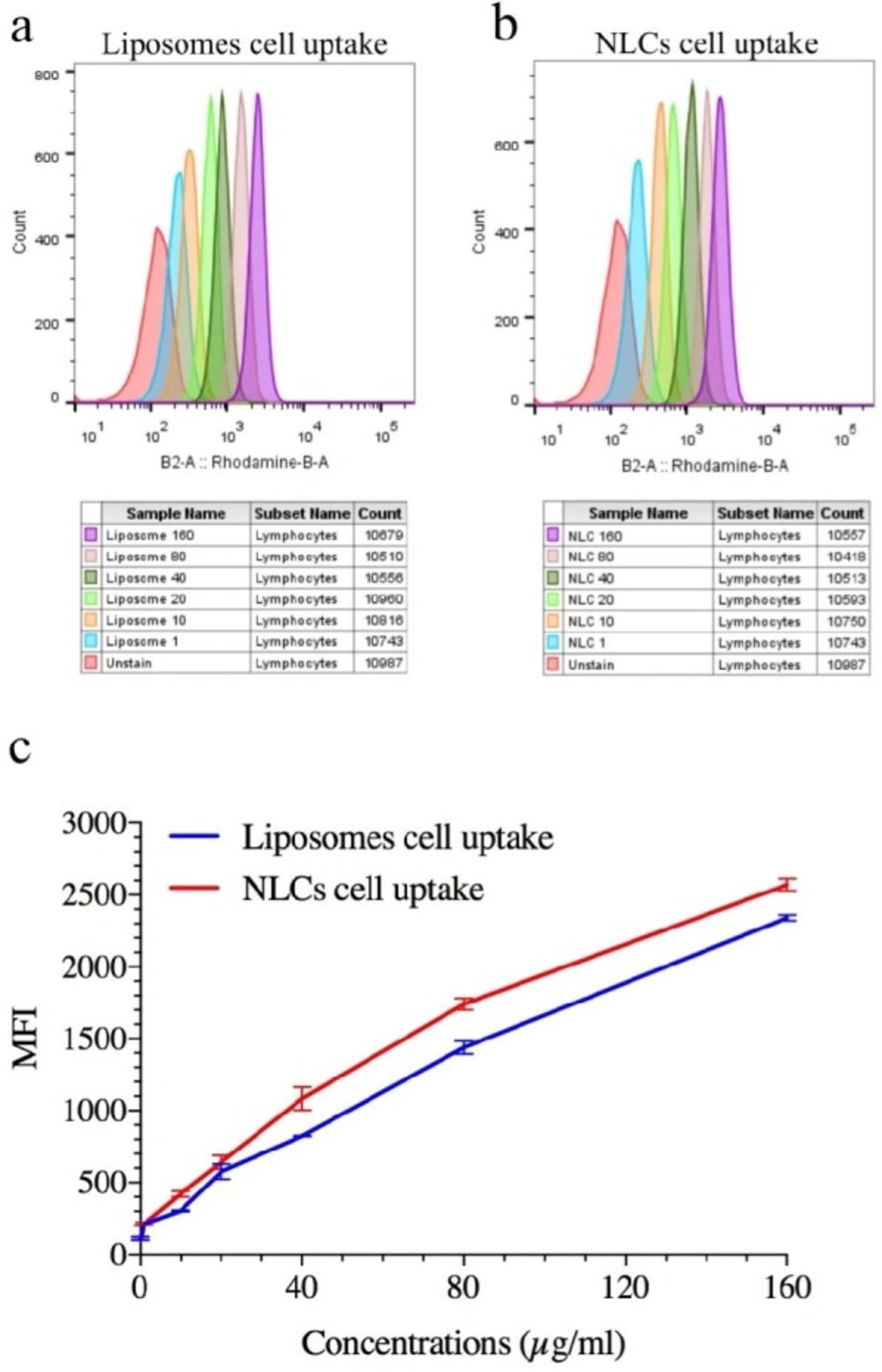
In-vitro cellular uptake
Rhodamine B (RhB) as a fluorescent agent moiety was employed to confirm cellular uptake of the prepared formulations by A549 cells. To this purpose, RhB was added to the NLC formulation (0.5% w/w rhodamine B ratio to lipid) in the melted lipid phase stage. After a complete mixing, the aqueous phase was added to it. The separation fluorescent of NLC from free florescent was done by using the Amicon tube (Ultra-30 kDa molecular weight cut-off membrane, Millipore, Germany). For this aim, the fluorescent NLCs were placed into the upper chamber of Amicon filter and centrifuged under 4k rpm for 15 min. This process was performed to ensure thoroughly elimination of un-entrapped rhodamine B from NLC formulation (
24). In the fluorescent - liposome purification, RdB was add to the liposome in construction of thin layer film step and encapsulated by liposome to mimic the encapsulation of the ELT. Next process of this purification has been done similar to the NLCs formulations (
25). A549 cells (5 x 10
4 per well) were seeded in six-well, after that the cells were treated with 1, 10, 20, 40, 80, and 160 µg/mL of RhB labeled NLCs and liposome formulations for 4h, when the cells reached to 70% confluency. The quantitative cellular uptake of the prepared formulations was measured by a flow cytometry.
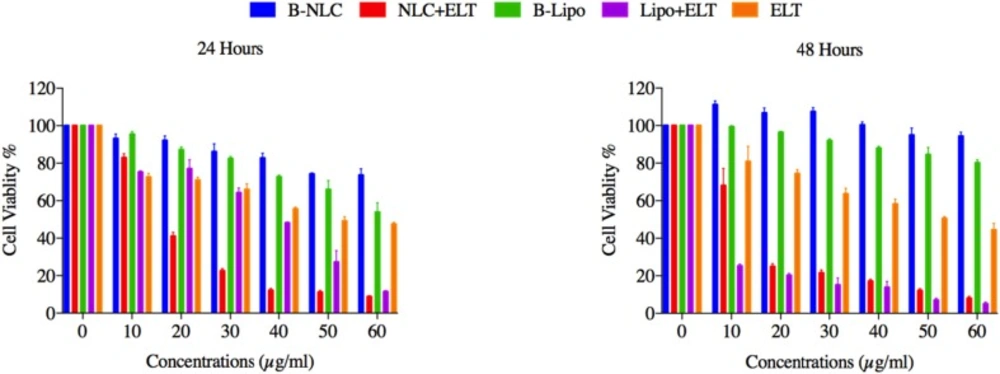
Cytotoxicity study
MTT (3-[4, 5-dimethyl-2-thiazolyl] - 2, 5 diphenyl tetrazolium bromide) assay was carried out to probe the effects of free ELT, ELT-NLCs, and ELT-liposome formulations on the viability of A549 cells (26). Briefly, 15×103 three time passaged A549 cells were seeded into each well of 96-well plate. Then, the cells were treated with different concentrations of free ELT (10-60 µg/mL) and its nano formulations. After 24 and 48 h, 50 mL of MTT solution (2mg/mL PBS) was added to each well. Incubated medium was replaced with 200 m L of DMSO after 4 h. Then, the absorption of the plates was read at 570nm by a TCAN Elisa Reader.
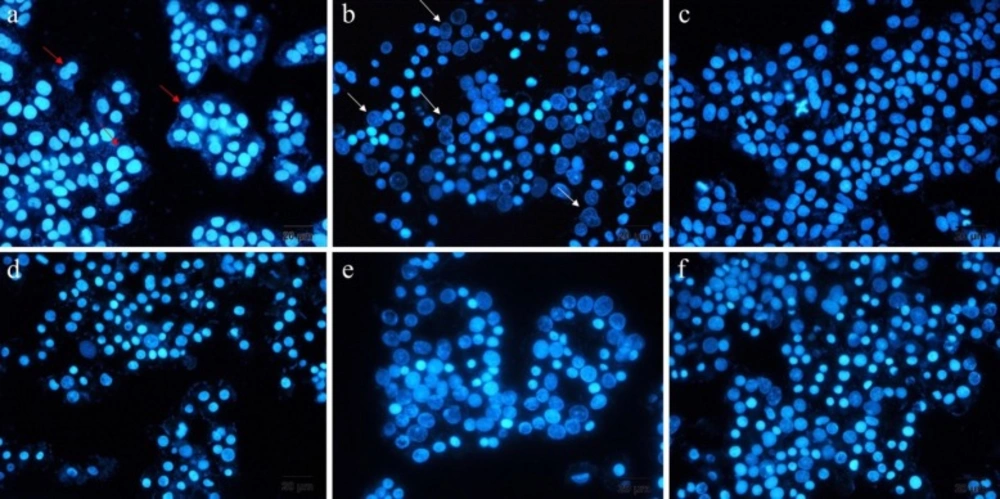
DAPI staining
DAPI staining is a method that represents the chromatin fragmentation. To implement this assay, A549 cells were seeded into six-well plates, including 12mm cover-slips at seeding density of 5 X 105 cells per well. The cultivated cells were treated with free ELT, blank NLCs and liposomes, ELT-liposome, ELT-NLCs, and docetaxel (300 µg/mL) as a positive control for 48 h. Then, the cells were fixed with 4% formaldehyde for 3 h. Following that, the cells were permeabilized with 0.1% Triton X-100 in PBS for 5 min. Next, the cells were stained with DAPI 0.1% for 10 Min. Finally, the studied cells were monitored by fluorescence imaging system (Cytation 5, Biotek, USA).
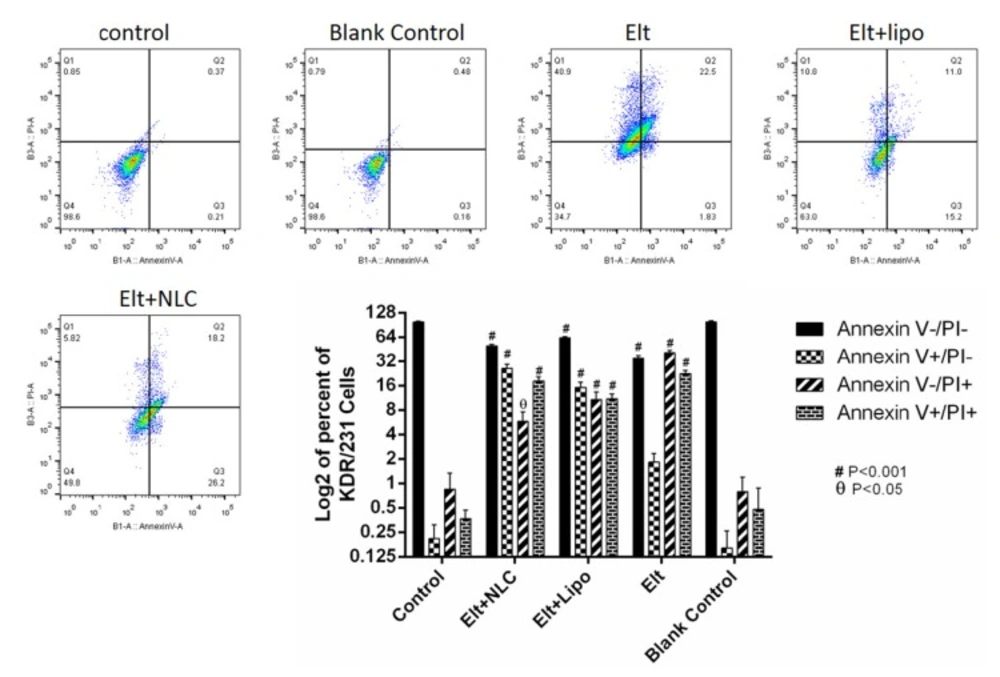
Flow cytometry
To measure the rate of cell apoptosis, A549 cells were seeded into six-well plate at a density of 5 × 105 cells per well in the RPMI 1640 media with 10% FBS. After 24 h, the media was changed with 2 mL fresh media, contained IC50 concentration of each treatment., and the cells were incubated for 48 h.
Following that, the cells were detached and stained via annexin V/PI apoptosis detection kit (Exbio, Czech Republic) according to the manufacture’s protocol. Then, the rate of apoptosis was analyzed by flow cytometer equipment (MACS Quant 10, Miltenyi Biotech GmbH). The achieved data was analyzed by using FlowJo software package (Treestar, Inc., San Carlos, CA) (
27).
Statistical analysis
All parameters were expressed as mean ± standard deviation. Statistical analyses were carried out using one-way and two-way analyses of variance (one-way and two-way ANOVA) with multiple comparisons between deposition data using a LSD significant difference test (Prism, version 7.0, Graphpad softwhere, INC). A P value less than 0.05 was considered as significant.