Compounds isolation and identification
The structure of isolated and identified compounds from
O. decumbens was depicted in
Figure 1 (
1-9).
Compound 1 was obtained as a colorless oil from the n-hexane fraction. Based on NMR spectrum (Figure S1a, in supplementary file) and ESI-MS analysis (Figure S1b) and compared with available data (26, 27), compound 1 was identified as octacosane (C28H58).
Compounds
2 and
3 were mixed together. According to the NMR spectrum of the mixture (Figure S2) in comparison with available data as well as TLC analysis comparing with available standards, it was identified to be the mixture of carvacrol and thymol (
28,
29).
Compound
4 was obtained as white crystals, identified as stigmasterol based on NMR spectra (Figure S3), and compared with available literature (
30).
Compound 5 was obtained as yellow crystals and identified as 3‐O‐β‐‐(6″‐O‐coumaroyl)glucopyranoside (tiliroside) according to 13H- and 13CNMR spectra as well as available data (31-33). 1HNMR (500 MHz, DMSO-d6): 8.02 (2H, d, J = 8.5 Hz, H-2’, H-6’), 7.59 (d, 1H, J = 16.0 Hz, H- γ), 7.54 ( 2H, d, J = 8.0 Hz, H-2’’, H-6’’), 6.89 (2H, d, J = 8.5Hz, H-3’, H-5’), 6.79 (2H, d, J = 8.0 Hz, H-3’’, H-5’’),6.40 (1H, d, J = 16.0 Hz, H-β), 6.40 ( 1H, s, H-8), 6.17 ( 1H, s, H-6), 5.75 ( 1H, d, J = 7.5 Hz, H-1’’), 3.35- 4.87 (6H, m, the rest of sugars protons). 13CNMR (125 MHz, DMSO-d6): 177.1 (C-4), 165.8 (C- α), 164.5 (C-7), 161.1 (C-5), 160.0 (C-4’’’), 159.8 (C-4’), 156.3 (C-2), 156.1 (C-9), 144.9 (C-γ), 132.5 (C-3), 130.7 (C-2’’’/C-6’’’), 130.3 (C-2’/C-6’), 125.1 (C-1’), 120.7 (C-1’’’), 115.8 (C- β), 115.1 (C-3’/C-5’) , 114.3 (C-3’’’/C-5’’’), 103.8 (C-10), 98.7 (C-1’’), 98.1 (C-6), 93.7 (C-8), 77.7 (C-2’’), 74.1 (C-3’’), 70.1(C-4’’), 60.6 (C-5’’), 48.9 (C-6’’)( Figures S4 & S5).
Compound
6 was obtained as yellow crystals. According to the following NMR characterization and comparing those data with the literature (
34) it was identified as kaempferol-3-O-(6’’-O-trans-coumaroyl) glucopyranoside 7-O- 6’’’’’ coumaroyl glucopyranoside.
1HNMR (500 MHz, DMSO-
d6): 8.05 (2H, d,
J = 8.0 Hz, H-2’, H-6’), 7.97 (2H, d,
J = 8.0 Hz, H-2’’, H-6’’), 7.62 (d, 1H,
J = 15.0 Hz, H- γ), 7.55 (2H, d,
J = 8.0 Hz, H-2’’’, H-6’’’’), 7.36 (d, 1H,
J = 15.0 Hz, H- γ´), 6.93 (2H, d,
J = 8.0 Hz, H-3’, H-5’), 6.87 (2H, d,
J = 8.0 Hz, H3’’’, H-5’’’’ ),6.80 (2H, d,
J = 8.0 Hz, H3’’’’, H-5’’’), 6.42 (1H, s, H-8 ), 6.40 (1H, d,
J = 15.0 Hz, H-β ), 6.17 ( 1H, d,
J = 15.0 Hz, H-β´), 6.13 ( 1H, s, H-6), 5.84 (1H, d,
J = 7.5 Hz, H-1’’’), 5.74 ( 1H, d,
J = 7.5 Hz, H-1’’), 3.35- 4.87 (the rest of sugars protons).
13C NMR
(125 MHz, DMSO-
d6): 177.1 (C-4), 166.1 (C-α), 165.8 (C-α´), 165.7 (C-7), 165.0 (C-4’’’’’), 164.3 (C-4’’’), 160.1 (C-5), 159.9 (C-4’), 159.9 (C-2), 156.4 (C-9), 145.2 (C- γ), 144.6 (C-γ´), 132.5 (C-3), 131.3 (C-2’’’, C-6’’’), 130.9 (C-2’, C-6’), 130.3 (C-2’’’’’, C-6’’’’’), 125.1 (C-1’), 125.0 (C-1’’’), 124.9 (C-1’’’’), 115.8 (C-β), 115.2 (C-β´), 115.1 ( C-3’, C-5’) , 114.3 (C-3’’’, C-5’’’), 113.8 (C-3’’’’’, C-5’’’’’), 103.9 (C-1’’’’), 103.8 (C-1’’), 98.3 (C-10), 98.2 (C-6), 93.7 (C-8), 75.2 (C-2’’’’), 74.6 (C-2’’), 74.3 (C-3’’’’), 73.9 (C-3’’), 71.9 (C-4’’’’), 71.4 (C-4’’), 71.0 (C-5’’’’’), 70.2 (C-5’’), 62.8 (C-6’’’’), 60.5 (C-6’’).
1H-
1H COSY spectrum showed connectivities among H-2’ and H-3’ as well as H-5’ and H-6’ on the B ring of kaempferol. Also, the correlation between H-2’’’ and H-3’’’, H-5’’’ and H-6’’’, H-2’’’’’ and H-3’’’’’, and H-5’’’’’’ and H-6’’’’’ on the aromatic rings of coumaroyl moieties attached to glucopyranoside group at C-3 and C-7 of kaempferol, respectively was demonstrated. It should be noted that desired correlations between olefinic protons (β /γ and β’ /γ ‘) of coumaroyl moieties were observed. Major NOSEY correlations showed that B ring protons of kaempferol correlated with protons of aromatic ring of cumaroyl moiety attached to glucopyranoside at C-3. In addition, there was a correlation between olefinic protons (β’/γ’) coumaroyl moiety attached to glucopyranoside at C-7 and H-8 (Figures S6-S9).
Compound
7 was identified as 3-hydroxythymol-6-
O-glucopyranoside according to data from NMR spectra and available literatures (
35,
36).
1HNMR (500 MHz, DMSO-
d6): 7.29 (1H, s, H-5), 6.93 (1H, s, H-2), 4.94 (1H, d,
J = 7.0 Hz, H-1´), 3.85-3.99 ( 1H, m, H-8), 3.35-4.11 (6 H, m, H-2´- 6’ ), 2.5 (3H, s, H-7), 1.53 (3H, d,
J = 6.8 Hz, H-9), 1.53 (3H, d,
J = 6.8 Hz, H-10).
13C NMR (125 MHz, DMSO-
d6): 148.9 (C-6), 147.4 (C-3), 132.1 (C-4), 121.3 (C-1), 119.1 (C-2), 111.8 (C-5), 102.8 (C-1’), 77.0 (C-2’), 76.9 (C-3’), 73.5 (C-4’), 70.1 (C-5’), 48.7 (C-6’), 25.6 (C-8), 23.1 (C-9), 22.6 (C-10), 15.8 (C-7) (Figures S10 and S11).
Compound
8 was identified as 6-hydroxythymol-3-
O-glucopyranoside based on data obtained from NMR spectra and literatures (
35,
37).
1HNMR (500 MHz, DMSO-
d6): 7.20 (1H, s, H-5), 7.00 (1H, s, H-2), 4.94 (1H, d,
J = 7.0 Hz, H-1’), 3.85-3.99 (1H, m, H-8), 3.35-4.11 (6H, m, H-2’- 6’), 2.50 (3H, s, H-7), 1.53 (3H, d,
J = 6.8 Hz, H-9), 1.53 (3H, d,
J = 6.8 Hz, H-10).
13C NMR (125 MHz, DMSO-
d6): 150.4 (C-6), 148.8 (C-3), 136.0 (C-4), 124.9 (C-1), 116.6 (C-2), 114.6 (C-5), 103.0 (C-1’), 77.0 (C-2’), 76.9 (C-3’), 73.6 (C-4’), 73.2 (C-5’), 60.9 (C-6’), 26.2 (C-8), 23.3 (C-9), 22.7 (C-10), 15.8 (C-7) (Figures S10 and S11).
Compound
9 was yield as yellow crystals. According to
1HNMR and
13CNMR spectra as well as data obtaining from literature, it was identified as kaempferol 3-Oβ -O-neohesperidoside-7-O-[2-O-(cis-feruloyl)] β -D-glucopyranoside (
34,
38,
39).
1HNMR (500 MHz, DMSO-
d6): 7.98 (2H, d,
J = 8.5 Hz, H-2’, H-6’ ), 7.85 ( 1H, s, H-2’’’’’), 7.52 (1H, d,
J = 11.0 Hz, H-γ), 6.91 (2H, d,
J = 8.5 Hz, H-3’, H-5’), 6.87 ( 1H, d,
J = 8.0 Hz, H-6’’’’’ ), 6.84 (1H, d,
J = 8.0 Hz, H-5’’’’’ ), 6.42 (1H, s, H-8 ), 6.39, (1H, d,
J = 11.0 Hz, H-β), 6.20 (1H, s, H-6), 5.44 (1H, d,
J = 7.0 Hz, H-1’’), 5.42 (1H, d,
J = 7.5 Hz, H-1’’’’), 5.08 (1H, bs, H-1’’’), 3.20-4.41 (the rest of sugars protons), 3.83 (3H, s, H-3’’’’’), 0.97 (3H, d,
J = 6.0 Hz, H-6’’’),.
13C NMR (125 MHz, DMSO-d6): 177.3 (C-4), 164.5 (C- α), 161.2 (C-7), 160.0 (C-5), 156.5 (C-4’), 156.4 (C-9), 149.4 (C-2), 148.4 (C-3’’’’’), 146.9 (C-4’’’’’), 144.7 (C-γ), 133.2 (C-3), 130.9 (C-6’’’’’133.0 (C-2’ , C-6’), 122.2 (C-1’’’’’), ) 121.0 (C-1’), 115.2 ( C-3’, C-5’), 115.1(C- β), 114.3 (C-5’’’’’), 113.3 (C-2’’’’’), 103.9 (C-10),101.2 (C-1’’’), 100.9 (C-1’’), 100.7 ( C-1’’’’), 98.8 (C-6), 93.8 (C-8), 76.4 (C-3’’’’), 75.9 (C-2’’), 75.7 (C-3’’), 74.3 (C-5’’), 74.1 (C-5’’’’), 71.7 (C-4’’’), 70.9 (C-2’’’’ ), 70.3 (C-2’’’), 70.2 (C-3’’’), 70.1 (C-4’’), 68.3 (C-5’’’), 68.2(C- 4’’’’), 66.9 (C-6’’), 66.8 (C-6’’’’ ), 55.6 (C3’’’’’). 17.7 (C-6’’’’) (Figures S12 and S13).
Anti-Helicobacter pylori and antibacterial activity
Anti
-Helicobacter pylori activity of total extract and different fractions were assessed using different concentrations in an agar dilution method (
Table 1)
. As shown in
Table 1, the
n-hexane fraction depicted the most significant anti
-H. pylori activity with the MIC value of 50 µg/mL. However, the other fractions showed much lower activity against
H. pylori (MICs = 1000, 1500, and 1750 µg/mL, respectively), and the total extract showed no inhibitory activity even at the concentration of 2000 µg/mL.
As reported in
Table 2, all fractions demonstrated antibacterial activity toward
S. aureus while all of them showed no antibacterial activity against
P. aeruginosa even at a concentration up to 500 µg/mL. It should be noted that the total extract and all fractions except ethyl acetate fraction depicted no antibacterial activity toward
S. epidermidis and
E. coli in the tested concentration range.
Urease inhibitory
The inhibitory activity of total extract and different fractions toward jack bean urease was reported in
Table 3. The best inhibitory activity was demonstrated by n-hexane (IC
50 = 285.44 µg/mL) and ethyl acetate (IC
50 = 285.06 µg/mL) fractions as compared to hydroxyurea as a positive control (IC
50 = 59.51 µg/mL). Chloroform and methanol fractions showed lower urease inhibitory activity with IC
50 values of 375.36 and 709.42 µg/mL, respectively. However, the total extract showed no inhibitory activity.
Moreover, the urease inhibitory of the isolated compounds as IC50 (mM) was evaluated (
Table 4)
. Among them, stigmasterol, 3‐
O‐β‐‐(6″‐
O‐coumaroyl), glucopyranoside (tiliroside), and carvacrol were found to be the most active as compared to positive control. It should be noted that thymol showed lower activity (IC50 = 1.33 mM) than its isomer carvacrol (IC50 = 0.70 mM). The mixture of hydroxyl thymol glucosides and compound
9 showed similar anti-urease activity (Inhibition percentage at 100 (µg/mL) concentration = 24.6%), and no activity was induced by compound
6 up to 250 µg/mL.
Molecular docking of the isolated compounds in the binding site of urease
Table 5 summarized all details related to the docking study of the isolated compounds in the binding site of urease. Stigmasterol, tiliroside, and carvacrol as the most active compounds inhibited the catalytic activities of urease through hydrogen bonding, metal/ion contact with Ni ions, and hydrophobic interactions. The interaction of the best-docked conformation of the ligand with active site residues is depicted in
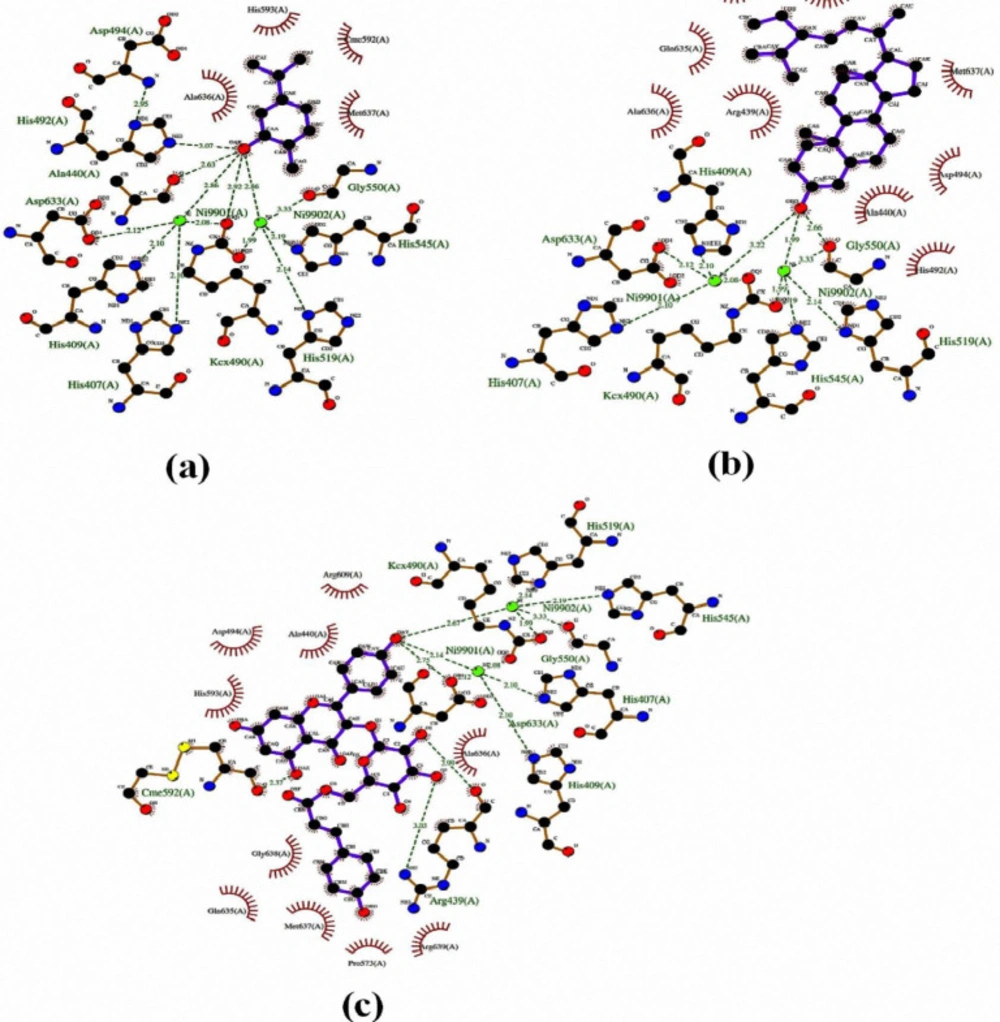
Figures 2 and S14-S18.
The hydroxyl groups of thymol and carvacrol interacted with the Ni ion and imidazole ring of His492 (
Table 5, Figure S14). In addition, the docked pose of carvacrol tightly engaged in nickel chelation along with several hydrogen bonds within the binding cavity (
Figure 2a). The hydroxyl group of cyclohexyl moiety of stigmasterol highlighted the formation of a bidentate bond with two-nickel ions (1.92 Å, 3.22 Å) and Gly550 (2.66 Å) (
Table 5). The substituted rings are deeply embedded into the hydrophobic pocket of the Ile411, Arg439, Ala440, His492, Asp494, Cme592, Gln635, Ala636, and Met637 (
Figure 2b).
Figure 2c shows that the hydroxyl groups of kaempferol moiety in the tiliroside were also engaged in the binding cavity through nickel ions (2.14 Å, 2.67 Å) hydrogen bonds with the oxygen atom of Asp633 and Cme592 side chains. In addition, both OH groups of glucoside moiety mediated through two hydrogen bonds with the side chain of Arg439. The lowest free binding energy of tiliroside was -8.49 kcal/mol with K
i of 598.78 nM. This is in good accordance with the experimentally observed value of urease inhibition percentage.
Similarly, compound
7 with glucopyranoside group at position 6 showed slightly better results than its counterpart (compound
8). The different OH groups of compound
7 formed hydrogen bonding and polar bonds with Arg439, Ala440, Kcx490, His492, Asp494, Gly550, Arg609, Asp633, and nickel ions (
Table 5, Figure S15). As shown in figure S16, the non-glucopyranoside moiety of compound 8 was observed more active than its homolog (compound 7) by interacting with Ni ions, Kcx490, His492, and His519. Four additional hydrogen bonds were also observed between oxygen’s glucopyranoside of compound
8 and the key residues, including Asp494, Cme592, and Ala636 (
Table 5). Compounds
6 and
9 have interacted with other residues shown in
Table 5 and Figures S17- S18.

![Structure of the isolated compounds from different fractions of <i>Oliveria decumbens </i>Vent. (a) Compound <b>1</b>: Octacosane, (b)Compound <b>2</b>: Carvacrol, (c) Compound <b>3</b>: Thymol, (d) Compound <b>4</b>: Stigmasterol, (e) Compound <b>5</b>: Kaempferol-3-O-(6''-O-trans-coumaryl) glucoside (Tiliroside), (f) Compound <b>6</b>: Kaempferol 3 -O-(6’’-O-trans-coumaryl)glucoside 7-O-(6’’’-O-trans-coumaryl) glucoside, (g) Compound <b>7</b>: 3-Hydroxythymol-6-O-D-Glucopyranoside, (h) Compound <b>8</b>: 6- Hydroxythymol-3-O-D-Glucopyranoside, (i) Compound <b>9</b>: Kaempferol 3-O-neohesperidoside-7-O-[2-O-(cis-feruloyl)]-D-glucopyranoside](https://brieflands.com/journals/ijpr/articles/127051/figures/ijpr-20-476-g001-preview.webp)