Wnt signaling pathway can be considered as a good target for developing drugs in order to control various types of cancers. Although several attempts have been done for designing drug candidates against Wnt signaling pathway, just a few of them have been successful in the clinical stage (
41).
Currently, several inhibitors have been detected which can target upstream or downstream proteins in this pathway leading to substantial anti-tumor activity. Porcupine inhibitors (IWPs) as small molecules inhibiting production of Wnt protein (
42), FJ9 inhibiting β-Catenin-destruction complex, 3289–8625 and NSC668036 targeting the FZD and DVL-PDZ interaction are some examples of these compounds (
43-
45). Monoclonal antibodies such as OMP-18R5 and OMP- 54F28 have been developed in this regard (
46). JW55 inhibiting PARP domain of tankyrase 1 and tankyrase 2, as well as JW67 and JW74 which suppress tumor growth in
Apc mutant mice, have been introduced as drug candidates too (
47,
48). Two most effective compounds, PFK115-584 and CGP049090 in a dose-dependent manner disrupt β-catenin/TCF complex (
49). It is suggested that upstream targeting of Wnt signaling pathway can be effective too. Wnt signaling pathway activation is highly dependent on the binding of Wnt ligands to Fzd receptors (
50,
51). It is well documented that Wnt ligands are the most important components of Wnt pathway. Blocking the binding of Wnt to Fzd receptor may suppress this pathway and increase apoptosis in cancer cells (
15,
52). Despite the important role of Wnt proteins in Wnt signaling pathway, inhibitors which can directly inhibit Wnt proteins or block interaction of Wnt-Fzd have not yet been reported. Hammad
et al. only designed small-molecule inhibitors against binging site 1 of Wnt4 (
53).
Studies have shown that Wnt2 protein is the main regulator of Wnt signaling pathway and its overexpression has been detected in various types of cancer, especially colorectal cancer (
54-
56). In this study, several compounds were identified against both binding sites of Wnt2 protein. MD simulations revealed that the compounds can efficiently bind and block Fzd-binding site of Wnt2.
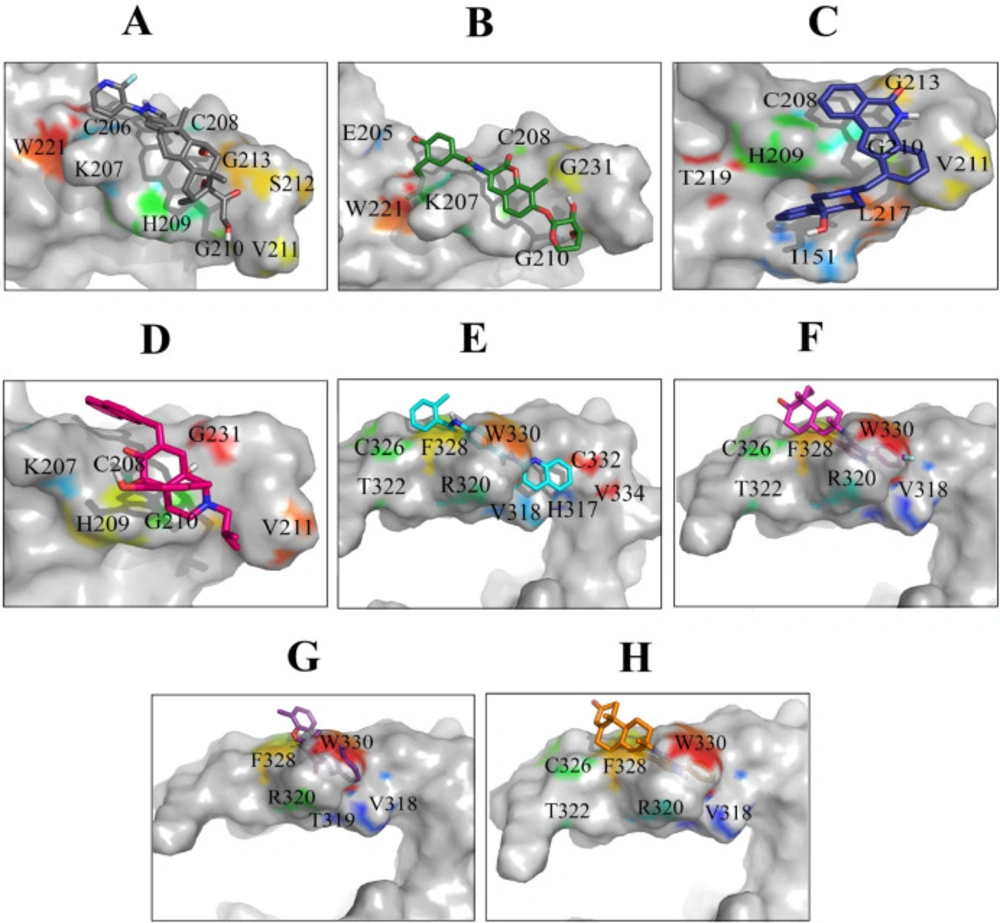
Most frequent protein–ligand atomic interactions are: hydrophobic interactions, hydrogen bonds, π-stacking and weak hydrogen bonds, salt bridges, amide stacking, and π- cation interactions (
57,
58). These interactions are well known and extensively applied to drug design. Hydrophobic interactions are the key motivating force in protein–drug interactions (
59). Therefore, lead compounds were investigated on the basis of the majority of the interactions with the binding sites of Wnt2 protein (
Figure 5). The result of protein-compound complexes showed that these inhibitors not only can interact with the residues located in the binding sites but also can interact with some other residues such as I
151, L
217, E
205, W
221 (binding site 1) and V
318, T
319, R
320, T
322, C
326, and C
332 (binding site 2).
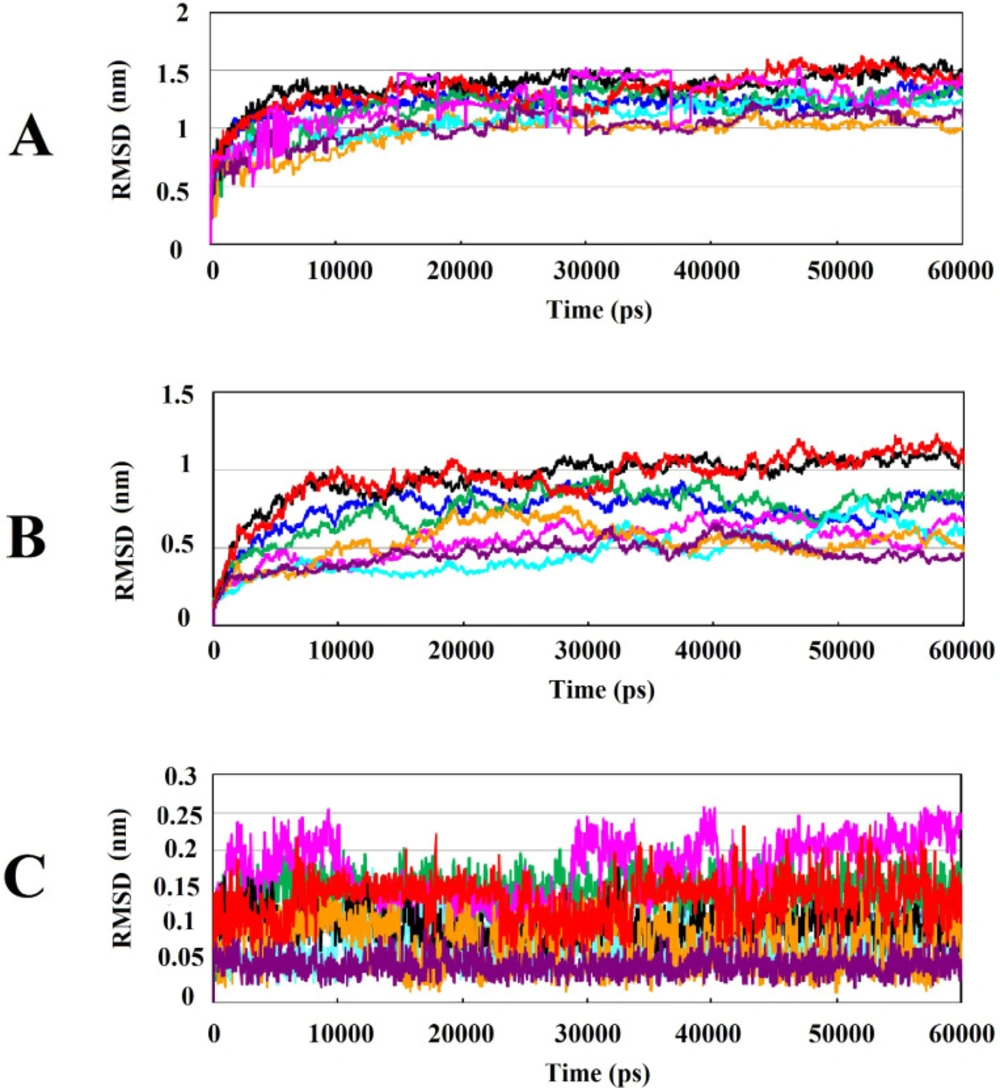
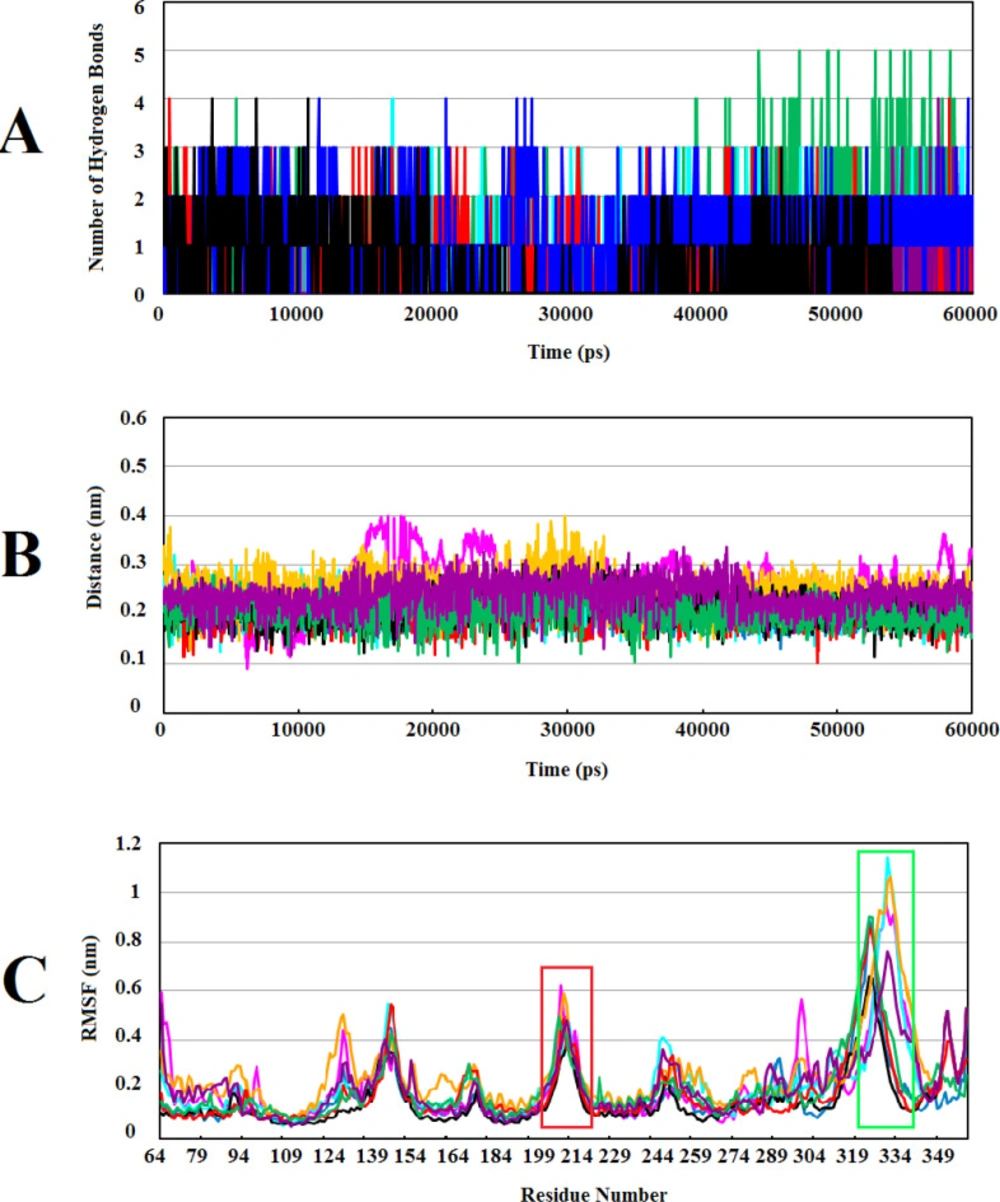
The most significant interactions between Wnt2 protein and the lead compounds were hydrophobic interactions. Hydrogen bonds were also contributed in the interactions between Wnt2 and compounds. The residues involved in hydrogen bonds were E205, C206, C208, H209, G210, V211 (binding site 1) and V318, R320 (binding site 2). In the binding site 1; ZINC40499329 compound revealed the highest binding affinity which was -9.7703 kcal/mol while its docking pose analysis showed four hydrogen bonds with C206, C208, G210 and G213 with the bond length of 2.68A˚, 2.48 A˚, 2.98 A˚, and 2.69 A˚, respectively. As well, ZINC60137214 compound in the binding site 2 showed the highest binding affinity of -11.53 kcal/mol. Different types of interactions; π- cation, hydrophobic interactions and a hydrogen bond with R320 residue with the bond length of 2.21A˚ were engaged in this binding which leads to the effective inhibition of Wnt2-Fzd receptor interaction. However, Wnt2-ZINC60137214 complex disclosed low RMSD which was almost stable in the last 30 ns.
It is well documented that methyl (CH3) groups affect the biological activity or binding affinity of small molecules. The presence of methyl (CH3) group in the small molecules makes them more hydrophobic and more prone for binding to proteins. Commonly, the methyl (CH3) groups interact with a hydrophobic pockets in the protein binding sites (
60,
61). Detailed analysis of the structure of ZINC71316775 and ZINC36221390 compounds against binding site 1 showed that they had a methyl (CH3) group which interacted with nonpolar residues of V
211 and W
221, respectively (Figures S1B and S1D, Supplementary File). Therefore, the presence of the methyl group in these compounds resulted in strong binding to Wnt2 protein. The RMSD evaluation of Wnt2-ZINC71316775 and Wnt2-ZINC36221390 complexes also confirmed that these compounds can efficiently bind to Wnt2.
In year of 2009, more than 40% of the small molecules which are in the clinical trial phase or under development contained an aromatic ring. Aromatic groups are involved in weak interactions and playing a key role in determining fundamental chemical and biochemical properties. On the other hand, π- Cation interaction is a non-covalent molecular interaction between positively charged amino acid side-chains: arginine, lysine, histidine and aromatic ring of ligands (
62). Arginine residue is more frequently involved in π-cation interactions than lysine side-chains (
59). During analysis of protein-compound interactions, we found that the selected lead compounds against the binding site 2 contained benzene rings which had π-cation interaction with R
320. Distances between R
320 and π-orbital of ZINC66078286, ZINC73408075, ZINC60137214, and ZINC06482373 were 4.14 A˚, 3.63 A˚, 3.52 A˚, and 3.54 A˚, respectively (Figures S1E-S1H, Supplementary File). Also, these compounds showed higher binding affinity than the selected lead compounds against binding site 1. Previous studies have documented the role of the π-cation interactions in drug design. While Sorafenib which forms a π-cation interaction with the lysine residue of the human p38 MAP kinase was approved for the treatment of liver cancer, lysine residue of ErbB4 kinase having π-cation interaction with lapatinib was approved for the treatment of breast cancer (63, 64). Therefore, the selected lead compounds against binding site 2 can be potential drug candidates for inhibiting Wnt signaling pathway.
Interestingly, we found that among selected lead compounds against binding site2, ZINC73408075 displayed different structural behavior and flexibility which was the highest fluctuation and instability in 40ns. Also, ZINC40499329 against binding site1 showed the highest RMSD value. Our investigation uncovered that they had a fluorine group in their structure.
S. Alapour
et al. have reported that the fluorine atom in the backbone of compounds generates higher non-conventional hydrogen bond interactions (inter and intra-molecular). These interactions probably can alter 3D structure of compounds and increase their flexibility (
65). Fluorine as the small and most highly electronegative element can interact directly with the target protein or indirectly impress the polarity of other groups of the compound. This would lead to the improvement in the binding affinity of the compound to the target protein (
66). Accordingly, ZINC40499329 and ZINC73408075 showed the highest fluctuations of RMSD in the free and complex form which may be due to the presence of fluorine atom upon phenyl ring (Figures S1A and S1F, Supplementary File). Also, they had high binding affinity to binding sites of Wnt2.
It has been demonstrated that the compounds with even a single fluorine atom can remarkably play a role in medicinal chemistry. Nowadays, many fluorinated compounds are synthesized in the pharmaceutical studies which are widely used in the treatment of diseases such as different types of cancer (
66,
67). As a result, ZINC73408075 and ZINC40499329 compounds can be promising drug candidates for inhibition of Wnt signaling pathway.
RMSF value of a protein is a significant index of many biological processes such as macromolecular recognition, protein activity, and complex formations. Atomic fluctuations in Wnt2-compound complexes were evaluated by computing RMSF values of each complex throughout MD simulations. Studies have shown that higher fluctuations are related to the amino acids in the loop structures and surface-exposed regions in the protein-protein interactions (
68,
69). S. Sikander Azam
et al. reported that residues at the edges of NTD and CTD domains of Wnt4 protein (128–158, 201–241 and 272–351 residues) had loop structures and showed more fluctuations than the other residues which destabilize them during MD simulations (
70). Accordingly, we found the highest fluctuations in the residues of binding site 1 and 2 (
Figure 4C). It seems that these flexible regions of human Wnt2 may have an important role in the interaction with Fzd CRD receptors. In our previous study, we investigated conformational changes in xWnt8 bound to Fzd8CRD during MD simulations. Our analysis uncovered that binding sites of xWnt8 have the highest rate of fluctuations (
17) . Based on these data and our results, theses flexible loop regions of binding sites of Wnt protein have a slight tendency to become rigid after binding to receptor or inhibitory compounds.
Gane
et al. demonstrated that the biological effects of compounds are associated with their chemical structure. Although the compounds with similar biological effects usually have identical structures, some compounds with dissimilar structures show comparable biological outcomes too (
71). Interestingly, we realized that ZINC73408075 and ZINC06482373 compounds have identical structure and their interactions with the residues in binding site2 of Wnt2 were alike too (Figures S1F and S1H, Supplementary File). Surprisingly they showed different structural behavior and flexibility during MD simulations (
Figures 3A and 3C). While alignment of their 2D structure disclosed that both have a similar core structure, the only difference was the presence fluorine atom which is attached to phenyl group in ZINC73408075.
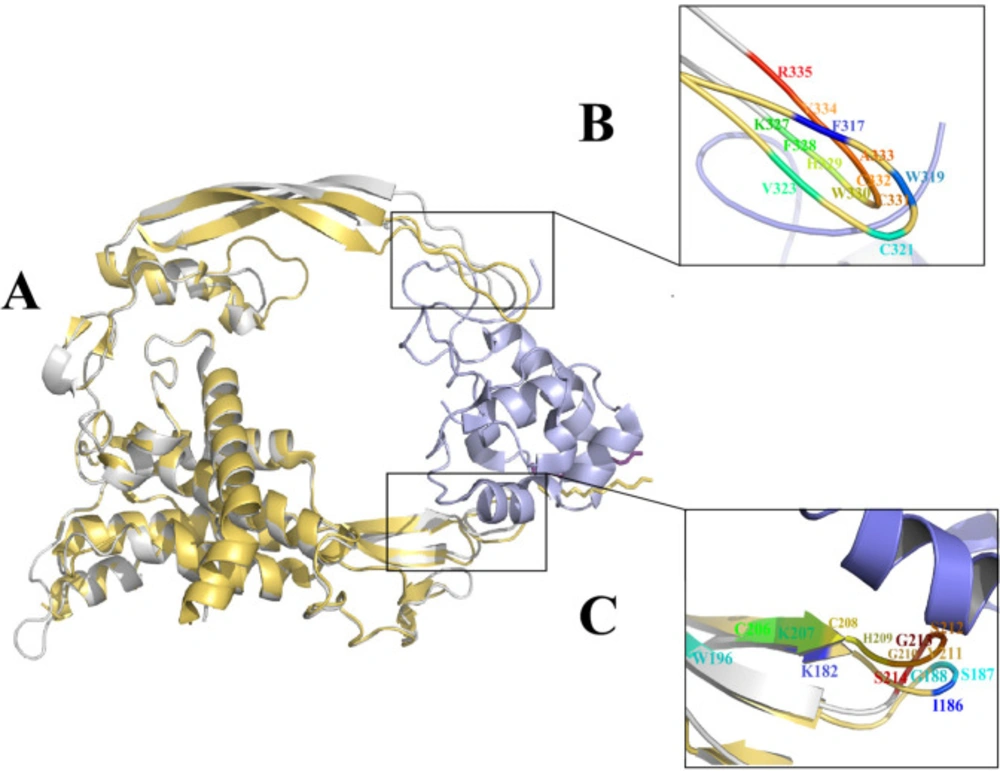
xWnt8-mFzd8 CRD and Wnt2 structure and evaluation of the binding sites on xWnt8 and Wnt2: (A) Structural alignment of the xWnt8 (yellow cartoon while Palmitoleic acid is shown sticks) and Wnt2 (gray cartoon while Palmitoleic acid is shown as purple sticks) in complex with mFzd8 CRD (light blue). (B) Close-up view of interacting residues of binding site 2 of xWnt8 and Wnt2 (C) Close-up view of interacting residues of binding site 1 of xWnt8 and Wnt2
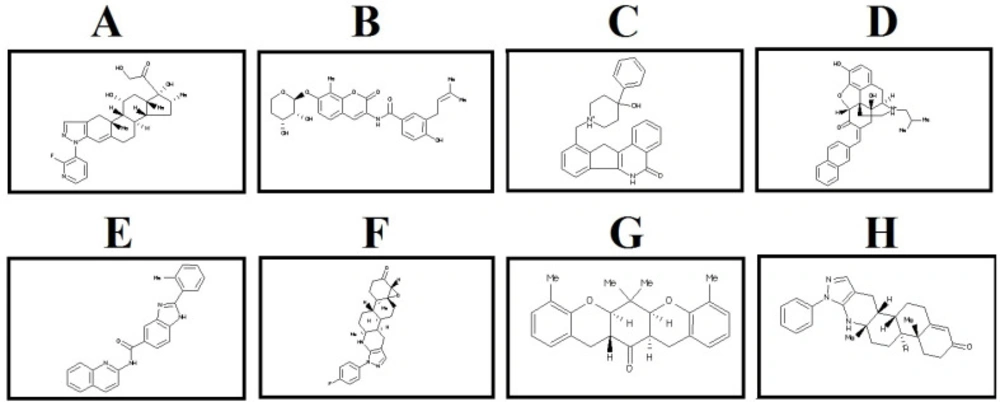
2D structure of the selected lead compounds from docking-based virtual screening. (A) ZINC40499329, (B) ZINC71316775, (C) ZINC35282053, (D) ZINC36221390 (E) ZINC66078286, (F) ZINC73408075, (G) ZINC60137214, (H) ZINC06482373
Analysis of RMSD value results during 60ns of simulations: (A) Root mean square deviation (RMSD) of backbone Cα atoms of the protein-compound complexes. (B) RMSD of backbone Cα atoms of Wnt2 in protein-compound complexes. (C) RMSD of backbone Cα atoms of compounds in protein-compound complexes. In all plots ZINC40499329, ZINC71316775, ZINC35282053, ZINC36221390, ZINC66078286, ZINC73408075, ZINC60137214, and ZINC06482373 are shown as black, green, blue, red, cyan, magenta, maroon and orang, respectively
Calculating the number of H-bonds, the minimum distance, and structural fluctuations in Wnt2-compound complexes during 60ns of MD simulations: (A) The number of H-bonds between the Wnt2 and compounds. (B) The minimum distances between Wnt2 and compounds. (C) Root Mean Square Fluctuation (RMSF) of backbone Cα atoms of Wnt2 within complexes versus residue’s number in the sequence, the binding sites 1 and 2 are shown in boxes with red and light green, respectively. In all plots ZINC40499329, ZINC71316775, ZINC35282053, ZINC36221390, ZINC66078286, ZINC73408075, ZINC60137214, and ZINC06482373 are indicated as black, green, blue, red, cyan, magenta, maroon and orang, respectively
Binding orientations of residues within the binding sites of Wnt2 during interaction with the selected lead compounds: Interactions of binding site 1 of Wnt2 with (A) ZINC40499329, (B) ZINC71316775, (C) ZINC35282053, (D) ZINC36221390 and binding site 2 of Wnt2 with (E) ZINC66078286, (F) ZINC73408075, (G) ZINC60137214, (H) ZINC06482373
| TUMj | MUTi | DLh | RBg | PSAf | HBDe | HBAd | CLSc | CLPb | MWa | Compound Name |
|---|
| BS1* |
|---|
| NR | NR*** | 4.8029 | 3 | 108.74 | 3 | 7 | -4.418 | 1.944 | 495.593 | ZINC40499329 |
| NR | NR | 3.5929 | 6 | 134.55 | 4 | 9 | -4.85 | 3.591 | 495.526 | ZINC71316775 |
| NR | NR | 2.4803 | 3 | 53.77 | 3 | 4 | -4.622 | 2.307 | 423.534 | ZINC35282053 |
| NR | NR | 1.6555 | 3 | 71.2 | 3 | 5 | -6.106 | 2.763 | 482.598 | ZINC36221390 |
| BS2** |
| NR | NR | 0.91752 | 3 | 70.67 | 2 | 5 | -6.059 | 5.061 | 378.434 | ZINC66078286 |
| NR | NR | 3.5501 | 1 | 59.45 | 1 | 5 | -5.495 | 2.941 | 435.541 | ZINC73408075 |
| NR | NR | 0.98722 | 0 | 35.53 | 0 | 3 | -5.361 | 4.952 | 362.46 | ZINC60137214 |
| NR | NR | 4.9035 | 1 | 46.92 | 1 | 4 | -5.285 | 3.857 | 401.552 | ZINC06482373 |
| Compound Name | binding affinity(kcal/mol) | HB-AAsa | NH-AAsb |
|---|
| BS1* | | | |
|---|
| ZINC40499329 | -9.77 | C206 ,C208, G210,G213 | K207, V211, H209,W221 |
| ZINC71316775 | -8.67 | E205, G213 | K207, C208, G210, W221 |
| ZINC35282053 | -8.48 | V211 | I151, C208, H209, G210, G213, L217, T219 |
| ZINC36221390 | -8.51 | G210, H209 | K207, C208, V211, G213 |
| BS2** | | | |
| ZINC66078286 | -10.91 | ___ | H317, , R320, T322, C326 , W330, F328, C332, V334 |
| ZINC73408075 | -11.09 | V318 | R320, T322, C32, F328, W330 |
| ZINC60137214 | -11.53 | R320 | V318, T319, , F328, W330 |
| ZINC06482373 | -11.44 | V318 | R320, T322 , C326, F328, W330 |