
Introduction
Experimental
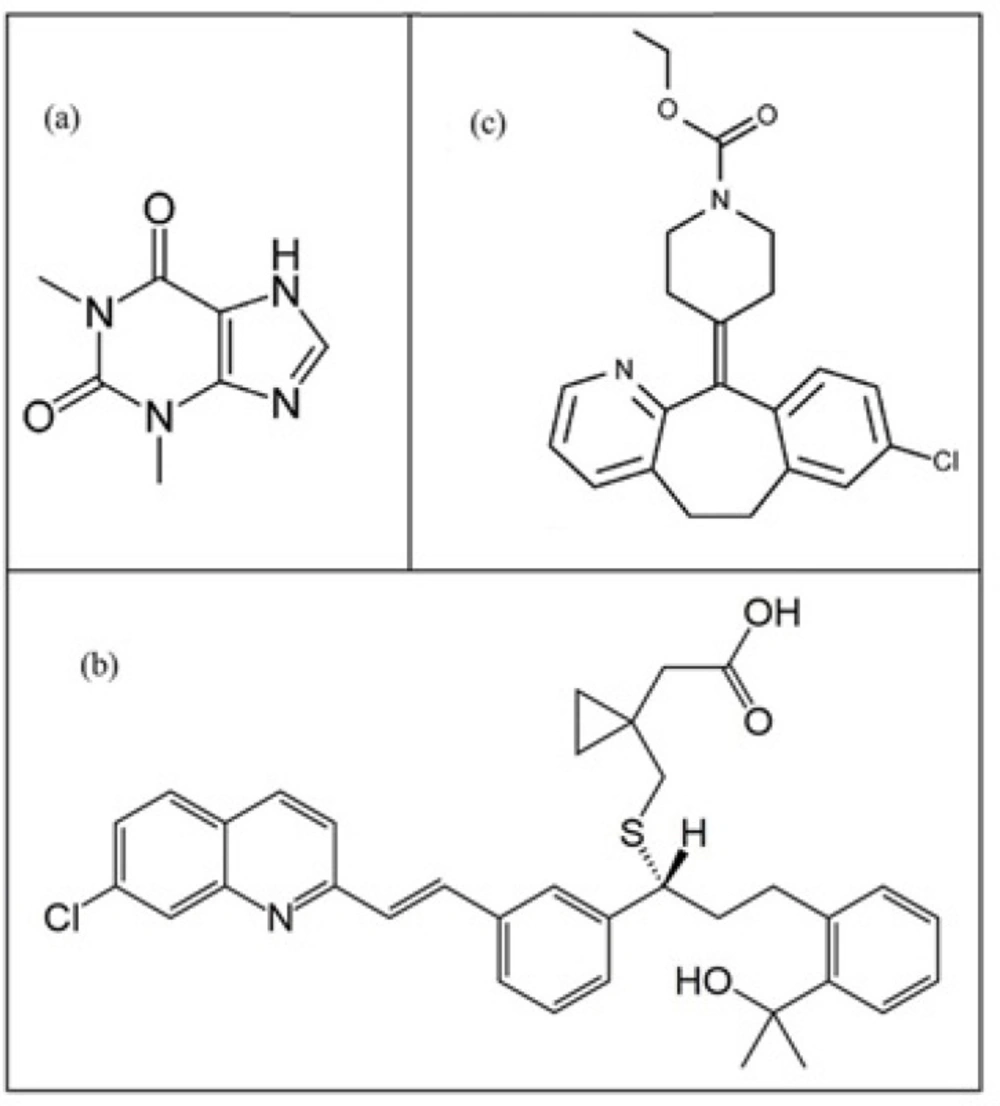
Molecular structure of three compounds in methanol: (a) theophylline, (b) montelukast and (c) loratadine.
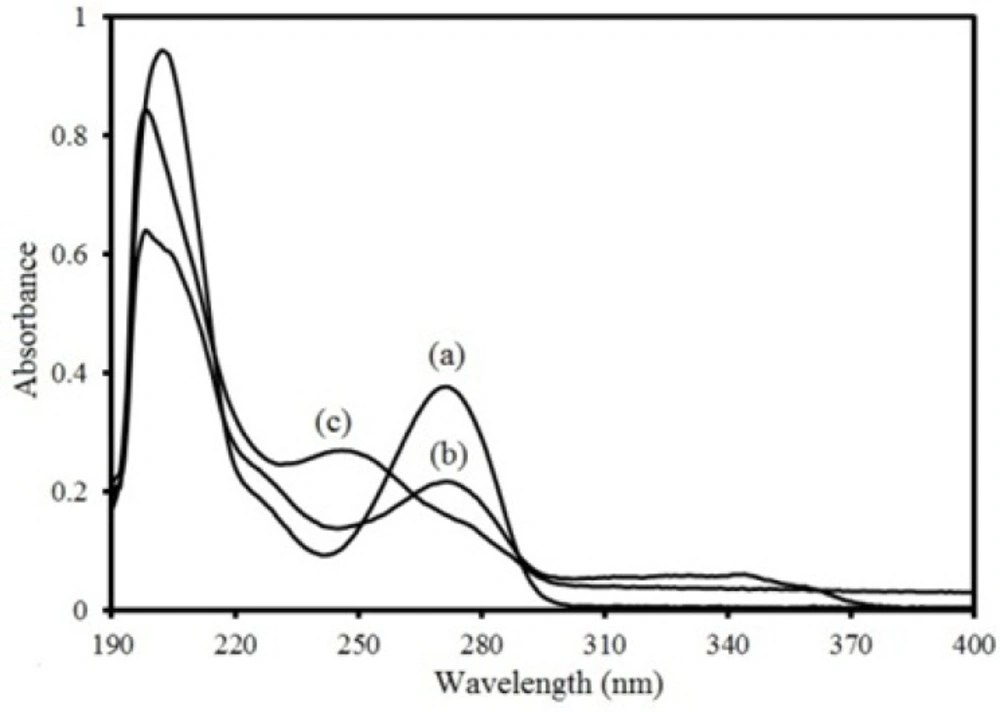
Electronic absorbance spectra of 6 ppm three compounds in methanol: (a) theophylline, (b) montelukast and (c) loratadine.
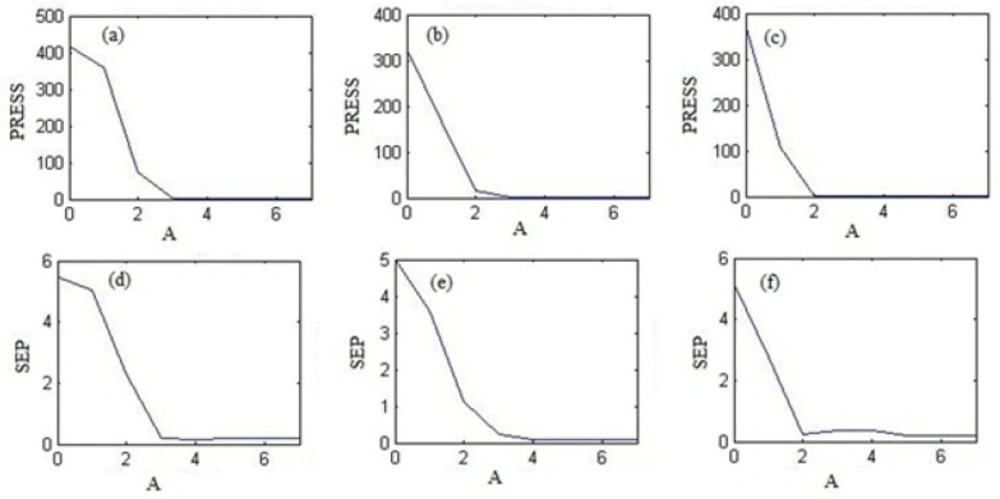
Variation of the PRESS as a function of the number of latent variables (A) (a) with PCR for theophylline, (b) with PLS1 for montelukast and (c) with HLA for loratadine. Plot of SEP vs. A (d) for theophylline by PCR regression, (e) for montelukast with PLS1 and (f) for loratadine with HLA.
| Analyte | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 | 13 | 14 | 15 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| THEO | 14 | 14 | 14 | 2 | 2 | 2 | 14 | 2 | 8 | 8 | 14 | 8 | 8 | 2 | 8 |
| MKST | 14 | 14 | 2 | 14 | 2 | 14 | 2 | 2 | 8 | 8 | 8 | 14 | 2 | 8 | 8 |
| LORA | 14 | 2 | 14 | 14 | 14 | 2 | 2 | 2 | 8 | 2 | 8 | 8 | 8 | 8 | 14 |
| PLS1 | PCR | HLA | PLS1 | PCR | |||
|---|---|---|---|---|---|---|---|
| Sample No. | Exp. | Pred. | RE | Pred. | RE | Pred. | RE |
| Test 1 | 11.00 | 10.54 | -4.18 | 10.53 | -4.27 | 10.56 | -4.00 |
| Test 2 | 11.00 | 10.72 | -2.54 | 10.72 | -2.54 | 10.72 | -2.54 |
| Test 3 | 11.00 | 10.82 | -1.64 | 10.83 | -1.54 | 10.84 | -1.45 |
| Test 4 | 3.00 | 2.97 | -1.00 | 2.98 | -0.67 | 2.93 | -2.33 |
| Test 5 | 3.00 | 2.98 | -0.67 | 2.99 | -0.33 | 2.93 | -2.33 |
| Test 6 | 3.00 | 2.87 | -4.33 | 2.87 | -4.33 | 2.83 | -5.67 |
| Test 7 | 11.00 | 10.86 | 1.27 | 10.87 | -1.18 | 10.93 | -0.66 |
| Test 8 | 3.00 | 3.03 | 1.00 | 3.04 | 1.33 | 3.06 | 2.00 |
| Test 9 | 7.00 | 7.14 | 2.00 | 7.14 | 2.00 | 7.13 | 1.86 |
| Test 10 | 7.00 | 7.02 | 0.28 | 7.01 | 0.14 | 7.13 | 1.86 |
| Test 11 | 11.00 | 11.01 | 0.09 | 11.01 | 0.09 | 11.08 | 0.73 |
| Test 12 | 7.00 | 6.91 | -1.28 | 6.90 | 1.43 | 6.94 | 0.86 |
| Test 13 | 7.00 | 7.20 | 2.85 | 7.20 | 2.86 | 7.25 | 3.57 |
| Test 14 | 3.00 | 3.21 | 7.00 | 3.18 | 6.00 | 3.14 | 4.67 |
| Test 15 | 7.00 | 7.01 | 0.14 | 6.98 | -0.28 | 6.58 | 6.00 |
| 2.02 | 1.93 | 2.70 |
is the mean of relative error percentage.
| PLS1 | PCR | HLA | |||||
|---|---|---|---|---|---|---|---|
| Sample No. | Exp. | Pred. | RE | Pred. | RE | Pred. | RE |
| Test 1 | 11.00 | 10.98 | -0.18 | 11.06 | 0.54 | 10.94 | -0.54 |
| Test 2 | 11.00 | 11.03 | 0.27 | 11.06 | 0.54 | 10.95 | -0.45 |
| Test 3 | 3.00 | 3.09 | 3.00 | 3.11 | 3.67 | 3.06 | 2.00 |
| Test 4 | 11.00 | 10.87 | -1.18 | 10.94 | -0.54 | 10.82 | -1.64 |
| Test 5 | 3.00 | 2.85 | -5.00 | 2.86 | -4.67 | 2.81 | -6.33 |
| Test 6 | 11.00 | 10.69 | -2.82 | 10.73 | -2.09 | 10.67 | -4.71 |
| Test 7 | 3.00 | 3.05 | 1.67 | 3.03 | 1.00 | 3.02 | 0.67 |
| Test 8 | 3.00 | 2.99 | -0.33 | 2.97 | -1.00 | 2.97 | -1.00 |
| Test 9 | 7.00 | 7.41 | 5.86 | 7.43 | 6.14 | 7.39 | 5.57 |
| Test 10 | 7.00 | 6.92 | 1.14 | 6.95 | -0.71 | 6.90 | -1.43 |
| Test 11 | 7.00 | 6.97 | -0.43 | 7.00 | 0.00 | 6.91 | -1.28 |
| Test 12 | 11.00 | 11.14 | 1.27 | 11.21 | 1.91 | 11.11 | 1.00 |
| Test 13 | 3.00 | 3.08 | 2.67 | 3.08 | 2.67 | 3.06 | 2.00 |
| Test 14 | 7.00 | 7.34 | 4.86 | 7.40 | 5.71 | 7.31 | 4.43 |
| Test 15 | 7.00 | 7.23 | 3.28 | 7.68 | 9.71 | 7.25 | 3.57 |
| 2.26 | 2.73 | 2.44 |
is the mean of relative error percentage.
| PLS1 | PCR | HLA | |||||
|---|---|---|---|---|---|---|---|
| Sample No. | Exp. | Pred. | RE | Pred. | RE | Pred. | RE |
| Test 1 | 11.00 | 11.44 | 4.00 | 11.45 | 4.09 | 11.39 | 3.54 |
| Test 2 | 3.00 | 3.15 | 5.00 | 3.15 | 5.00 | 3.01 | 0.33 |
| Test 3 | 11.00 | 11.22 | 2.00 | 11.22 | 2.00 | 11.19 | 1.73 |
| Test 4 | 11.00 | 11.50 | 4.54 | 11.51 | 4.64 | 11.50 | 4.54 |
| Test 5 | 11.00 | 11.37 | 3.36 | 11.37 | 3.36 | 11.35 | 3.18 |
| Test 6 | 3.00 | 3.19 | 6.33 | 3.20 | 6.67 | 3.09 | 3.00 |
| Test 7 | 3.00 | 2.94 | 2.00 | 2.94 | 2.00 | 2.90 | 3.33 |
| Test 8 | 3.00 | 3.00 | 0.00 | 2.99 | 0.33 | 3.03 | 1.00 |
| Test 9 | 7.00 | 7.37 | 5.28 | 7.37 | 5.28 | 7.28 | 4.00 |
| Test 10 | 3.00 | 3.10 | 3.33 | 3.11 | 3.67 | 3.13 | 4.33 |
| Test 11 | 7.00 | 7.17 | 2.43 | 7.17 | 2.43 | 7.05 | 0.71 |
| Test 12 | 7.00 | 7.24 | 3.43 | 7.25 | 3.57 | 7.23 | 3.28 |
| Test 13 | 7.00 | 7.26 | 3.71 | 7.26 | 3.71 | 7.25 | 3.57 |
| Test 14 | 7.00 | 7.27 | 3.86 | 7.28 | 4.00 | 7.25 | 3.57 |
| Test 15 | 11.00 | 11.36 | 3.27 | 11.41 | 3.73 | 11.17 | 1.54 |
| 3.50 | 3.63 | 2.78 |
is the mean of relative error percentage.
| THEO | MKST | LORA | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Parameters | PLS1 | PCR | HLA | PLS1 | PCR | HLA | PLS1 | PCR | HLA |
| Aa | 4 | 4 | 13 | 5 | 5 | 7 | 4 | 4 | 6 |
| R2CV | 0.998 | 0.998 | 0.998 | 0.999 | 0.999 | 0.999 | 0.998 | 0.998 | 0.998 |
| SEPCV | 0.173 | 0.170 | 0.174 | 0.081 | 0.091 | 0.085 | 0.173 | 0.195 | 0.174 |
| R2PRED | 0.996 | 0.997 | 0.995 | 0.994 | 0.990 | 0.993 | 0.991 | 0.990 | 0.993 |
A is the number of factor or latent variable and obtained at minimum prediction residual error sum of squares (PRESS)56.
| THEO | MKST | LORA | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Parameters | PLS1 | PCR | HLA | PLS1 | PCR | HLA | PLS1 | PCR | HLA |
| SEN | 0.119 | 0.127 | 0.018 | 0.062 | 0.069 | 0.040 | 0.081 | 0.083 | 0.022 |
| SEL | 0.464 | 0.498 | 0.072 | 0.178 | 0.198 | 0.114 | 0.342 | 0.350 | 0.094 |
| LOD | 0.03 | 0.03 | 0.18 | 0.06 | 0.05 | 0.09 | 0.05 | 0.05 | 0.10 |
| γ −1 | 0.010 | 0.010 | 0.059 | 0.020 | 0.016 | 0.030 | 0.016 | 0.016 | 0.033 |
| Sample | This work | LOD (Ref.) | ||||||
|---|---|---|---|---|---|---|---|---|
| THEO | 0.03 | 0.36 (14) | 0.3 (12) | 0.18 (9) | 0.072 (15) | 0.067 (16) | 0.03 (18) | 0.01 (4) |
| MKST | 0.05 | 0.293 (21) | 0.2 (28) | 0.1 (25) | 0.075 (29) | 0.009 (26) | _ | _ |
| LORA | 0.05 | 0.16 (30) | 0.048 (39) | 5.0×10-4 (36) | 2.5×10-4 (33) | 1.2×10-5 (38) | _ | _ |
| Predicted | ||||
|---|---|---|---|---|
| Sample No. | Actual | PLS1 | PCR | HLA |
| Art 1a | 3.00 | 2.99±0.09 (99.67) b | 2.99±0.07 (99.67) | 3.07±0.06 (102.33) |
| Art 2 | 4.00 | 4.14±0.13 (103.50) | 4.05±0.08 (101.25) | 4.14±0.08 (103.50) |
| Art 3 | 6.00 | 6.02±0.06 (100.33) | 6.01±0.05 (100.16) | 6.04±0.05 (100.67) |
| Art 4 | 8.00 | 8.07±0.08 (100.87) | 8.02±0.06 (100.25) | 7.95±0.07 (99.37) |
| Art 5 | 9.00 | 9.06±0.09 (100.67) | 9.05±0.08 (100.55) | 9.13± 0.09 (101.44) |
| Art 6 | 0.00 | –0.05±0.06 (–) | −0.05±0.05 (–) | −0.04±0.05 (–) |
| Art 7 | 8.00 | 8.12±0.10 (101.50) | 8.07±0.09 (100.87) | 7.92±0.08 (99.00) |
| Art 8 | 0.00 | –0.06±0.06 (–) | −0.07± 0.06 (−) | −0.05±0.07 (–) |
| 101.33 | 101.32 | 101.97 |
Art is artificial samples and Unk is unknown samples.
The reported values of standard deviations (±S.D.) are obtained from four replicates. Recovery percentages are shown in parentheses.
| Predicted | ||||
|---|---|---|---|---|
| Sample No. | Actual | PLS1 | PCR | HLA |
| Art 1a | 4.00 | 4.06±0.07 (101.50) b | 4.06±0.07 (101.50) | 4.12±0.11 (103.00) |
| Art 2 | 5.00 | 5.02±0.17 (100.40) | 5.15±0.17 (103.00) | 5.03±0.14 (100.60) |
| Art 3 | 7.00 | 6.90±0.09 (98.57) | 6.92±0.08 (98.86) | 6.85±0.13 (97.85) |
| Art 4 | 10.00 | 9.97±0.10 (99.70) | 9.85±0.14 (98.50) | 10.00±0.10 (100.00) |
| Art 5 | 5.00 | 5.18±0.12 (103.60) | 5.20±0.19 (103.80) | 4.86±0.16 (97.20) |
| Art 6 | 10.00 | 10.16±0.12 (101.60) | 10.16±.012 (101.60) | 10.14±0.12 (101.40) |
| Art 7 | 0.00 | –0.02±0.03 (–) | −0.02±0.04 (−) | −0.03±0.12 (–) |
| Art 8 | 0.00 | –0.03±0.03 (–) | −0.01± 0.04 (−) | −0.05±0.13 (–) |
| 99.96 | 101.40 | 99.21 |
Art is artificial samples and Unk is unknown samples.
The reported values of standard deviations (±S.D.) are obtained from four replicates. Recovery percentages are shown in parentheses.
| Predicted | ||||
|---|---|---|---|---|
| Sample No. | Actual | PLS1 | PCR | HLA |
| Art 1a | 4.00 | 4.08±0.13 (102.00) b | 4.08±0.13 (102.00) | 4.06±0.21 (101.50) |
| Art 2 | 5.00 | 5.13±0.14 (102.60) | 5.17±0.13 (103.40) | 5.10±0.22 (102.00) |
| Art 3 | 9.00 | 8.96±.012 (99.56) | 9.10±0.09 (101.11) | 8.72±0.16 (96.89) |
| Art 4 | 10.00 | 10.08±0.14 (100.80) | 10.08±0.14 (100.80) | 10.02±0.21 (100.20) |
| Art 5 | 13.00 | 12.85±0.10 (98.84) | 12.85±0.10 (98.84) | 13.05±0.22 (100.38) |
| Art 6 | 10.00 | 10.29±0.09 (102.90) | 10.28±.012 (102.80) | 9.95±0.17 (99.50) |
| Art 7 | 4.00 | 4.02±0.14 (100.50) | 4.12±0.11 (103.00) | 4.00±0.13 (100.00) |
| Art 8 | 0.00 | –0.12±0.10 (–) | −0.17±0.15 (−) | −0.05±0.13 (–) |
| 101.82 | 102.12 | 100.71 |
Art is artificial samples and Unk is unknown samples.
The reported values of standard deviations (±S.D.) are obtained from four replicates. Recovery percentages are shown in parentheses.
