Although neurodegeneration in MS was described by Charcot, the pioneering introducer of the disease in 1868, there is no approved medication mechanistically designed on the basis of this pathological event. In recent decades, monitoring of disability progression in patients treated with approved medications has shown the need for better medications which can protect the cells and axons in the CNS (
32). Therefore, finding neuroprotective agents to slow down or prevent disability progression has become a priority in MS research. Novel agents targeting the neurodegenerative process in MS are being examined in MS models (
32). One group of these neuroprotective agents interrupt the pathophysiolical processes involved in cell death and axonal loss (
4). Excitotoxicity has been associated with axonal loss and cell death in many acute neurological insults and chronic neurodegenerative states including MS (
4,
8-
10). The effect of excitotoxicity initiates from the first days of the disease course in MS (
10). Glutamate is considerably elevated in the CNS. Elevated concentration of glutamate leads to activation of different ionotropic receptors opens their ion channels and significantly increases the influx of calcium ions into the cells. High concentration of intracellular calcium induces activation of many enzymes and signaling cascades which might cause neuronal cell death and axonal loss (
11,
12). It has been proposed that neuronal injury is more efficiently triggered when calcium ions enter neurons at specific entry points (
11,
13,
14). According to this hypothesis, NMDARs are major routs of calcium influx in glutamate excitotoxicity causing prolonged calcium build-up in the neural cells and cell death. NMDAR consists of an NR1 subunit combined with a variety of NR2A to NR2D and NR3 subunits. During the course of EAE, NR2A and NR2B subunits have been reported to up-regulate in CNS tissues (
33,
34). Cell death might be a consequence of activation of NR2B-containing NMDARs by glutamate during excitotoxicity (
19). In contrast to NR2B, activation of NR2A-containing NMDARs can protect the cells by transferring pro-survival signals (
21). Calcium-dependent cell injury can be modulated by selective inhibition of NR2B-containing NMDARs without inhibition of NR2A-containing NMDARs which probably protects the cell during excitotoxicity. Neuroprotection by some medications has been attributed to inhibition of NR2B-containing NMDARs (
35). To find more effective neuroprotective agents with fewer adverse effects, NR2B-containing NMDARs antagonists have been tested in some diseases (
21,
36). In MS, however, only non-selective NMDAR antagonists have been evaluated.
In this study the effect of inhibition of the NR2B-containing NMDARs was evaluated in EAE using a highly selective and potent antagonist of this receptor subtype, RO 25-6981 (
23). RO 25
-6981 is a potent and selective blocker of NMDARs containing the NR2B subunit. The selectivity of Ro 25-6981 for inhibition of NR2B-containing NMDARs is 5000-fold more than inhibition of NR2A-containing NMDARs. In the first step of a preclinical drug screening study, several doses of this antagonist were compared with the vehicle (PBS) as negative control and memantine, a non-selective antagonist of NMDAR as positive control. As menantine shows therapeutic effects in acute EAE by inhibition of NMDARs, we used it as a control drug in this study (
37). Memantine is a clinically prescribed NMDAR antagonist under clinical trials on several neurodegenerative diseases or acute CNS catastrophes; however, the search for new and more potent NMDAR modulators with greater efficacy is ongoing (38). In animal models of MS memantine has been effective in abrogation of neurological deficits and modulation of the disease course when administered either prophylactically (concurrent with induction of EAE) or therapeutically (just after the onset of the disease) (
17,
18,
37). Menantine has been studied in clinical phases II and III of several clinical trials related to MS. Currently, memantine is under further MS trials (
39-
42).
In our study, the therapeutic administration of drugs was started when initial signs were seen in EAE-induced mice. A non-induced group was also monitored continously to compare the changes with induced groups.
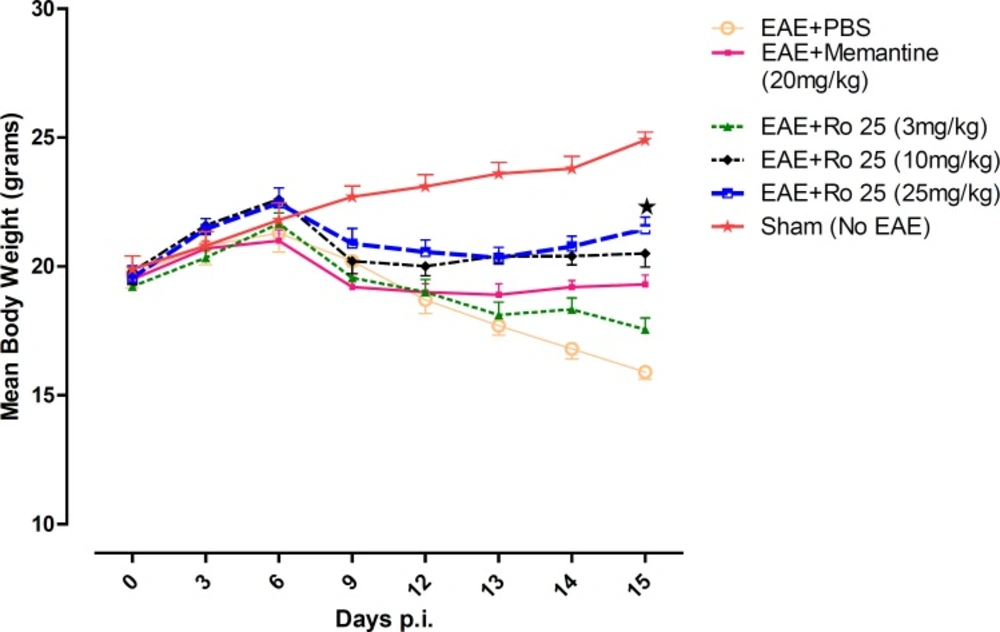
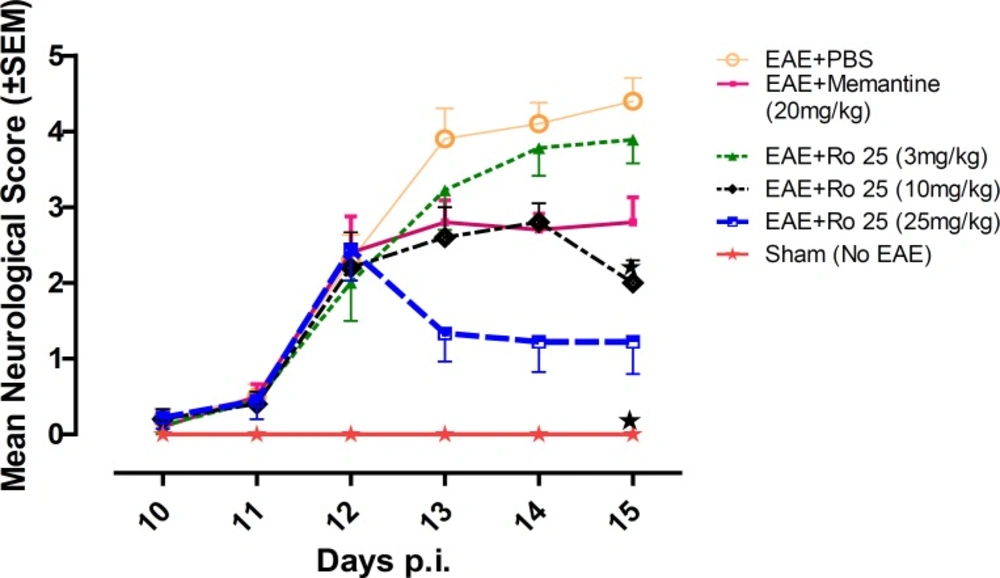
In our study, it was found that pharmacological inhibition of NR2B-containing NMDARs may have beneficial effects in modulation of neurological deficits. In terms of suppression of clinical disease progression, the therapeutic effect of RO 25-6981 when administered with the highest dose was more than all other experimental drugs including memantine. Moderate dose of RO 25-6981 was also effective in disease suppression. The extent of disease suppression by this dose was less than the high dose, although considering the Bonferroni's correction, the difference was not significant. The weight was also less decreased with administration of RO 25-6981. EAE mice start progressive weight loss from day 6 p.i. All groups showed a similar trend of weight loss till day 12, but groups under treatment with RO 25-6981, especially group 5 (treated with 25 mg/Kg/day) displayed gradual recovery of weight from day 12 p.i. This effect was dose dependent. On day 15, the mean weight in groups was significantly different. High-dose RO 25-6981 was more effective in this regard.
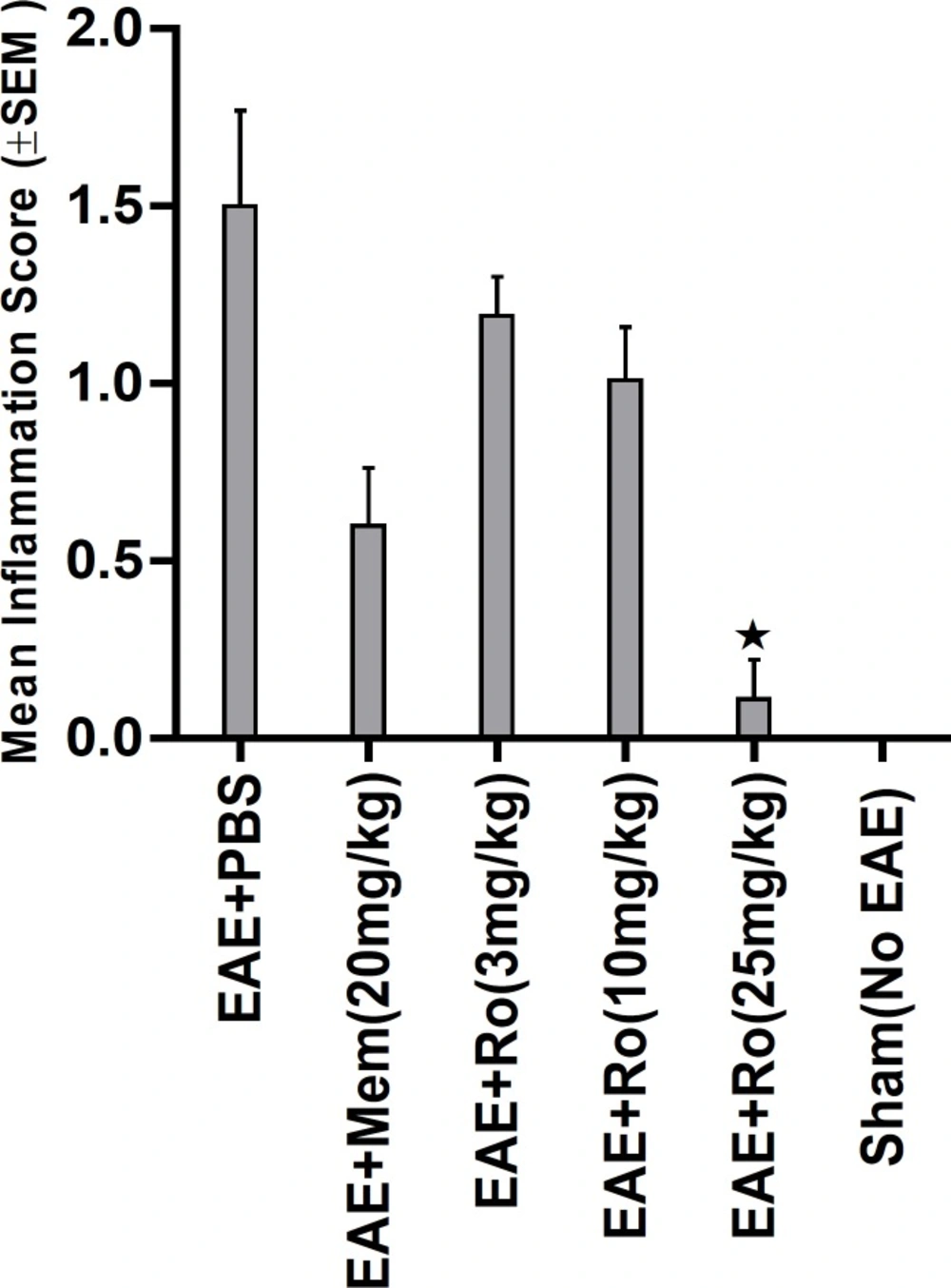
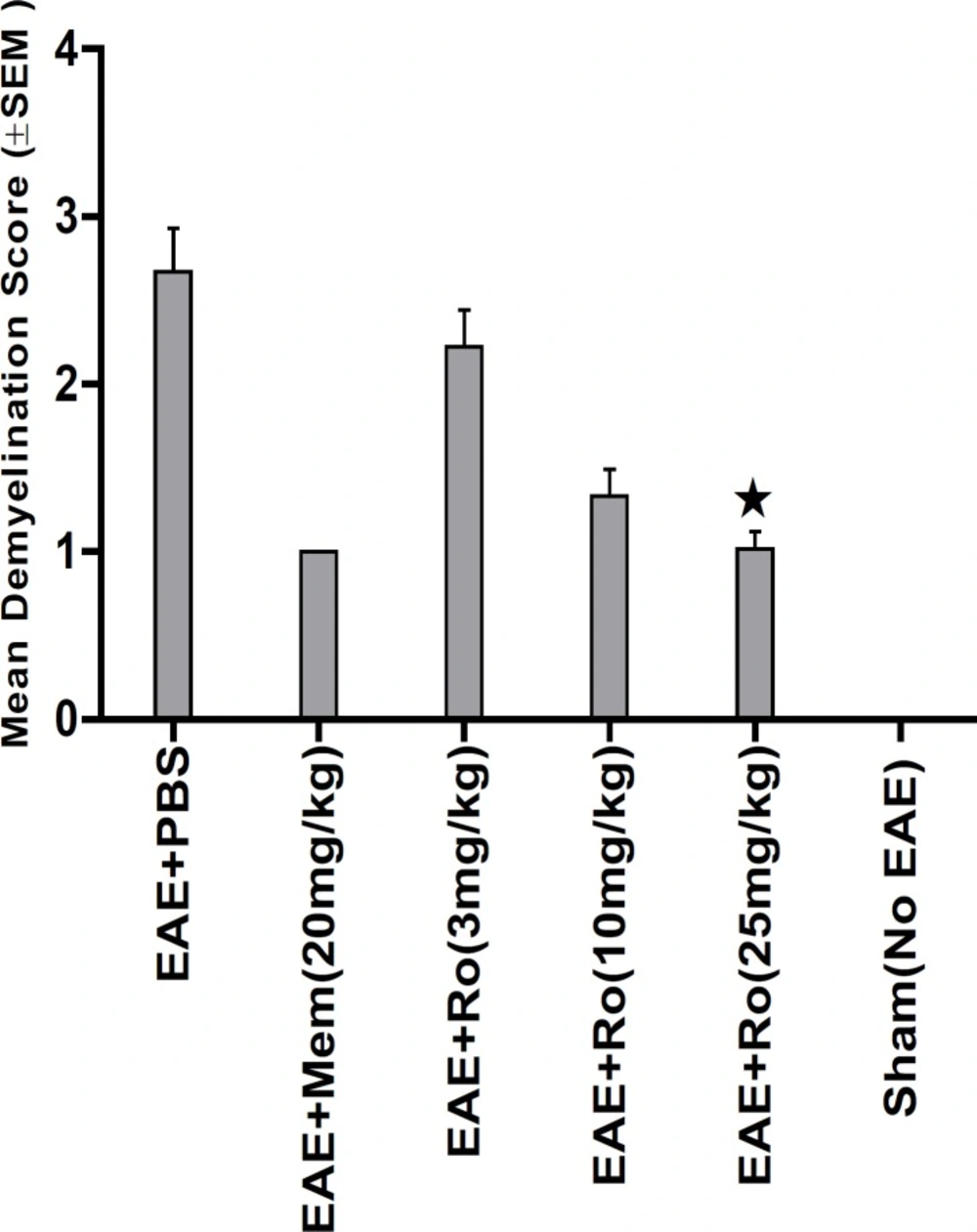
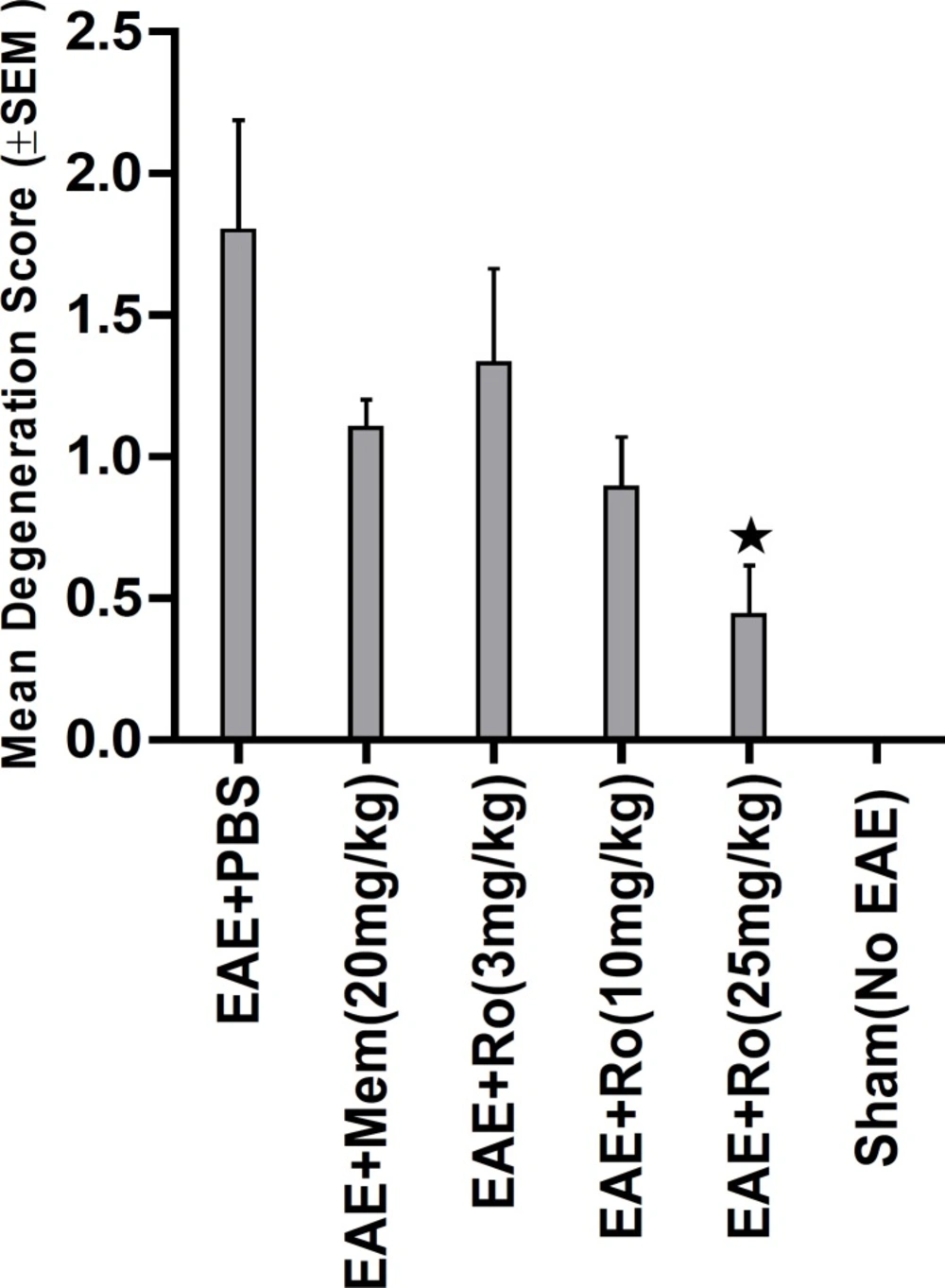
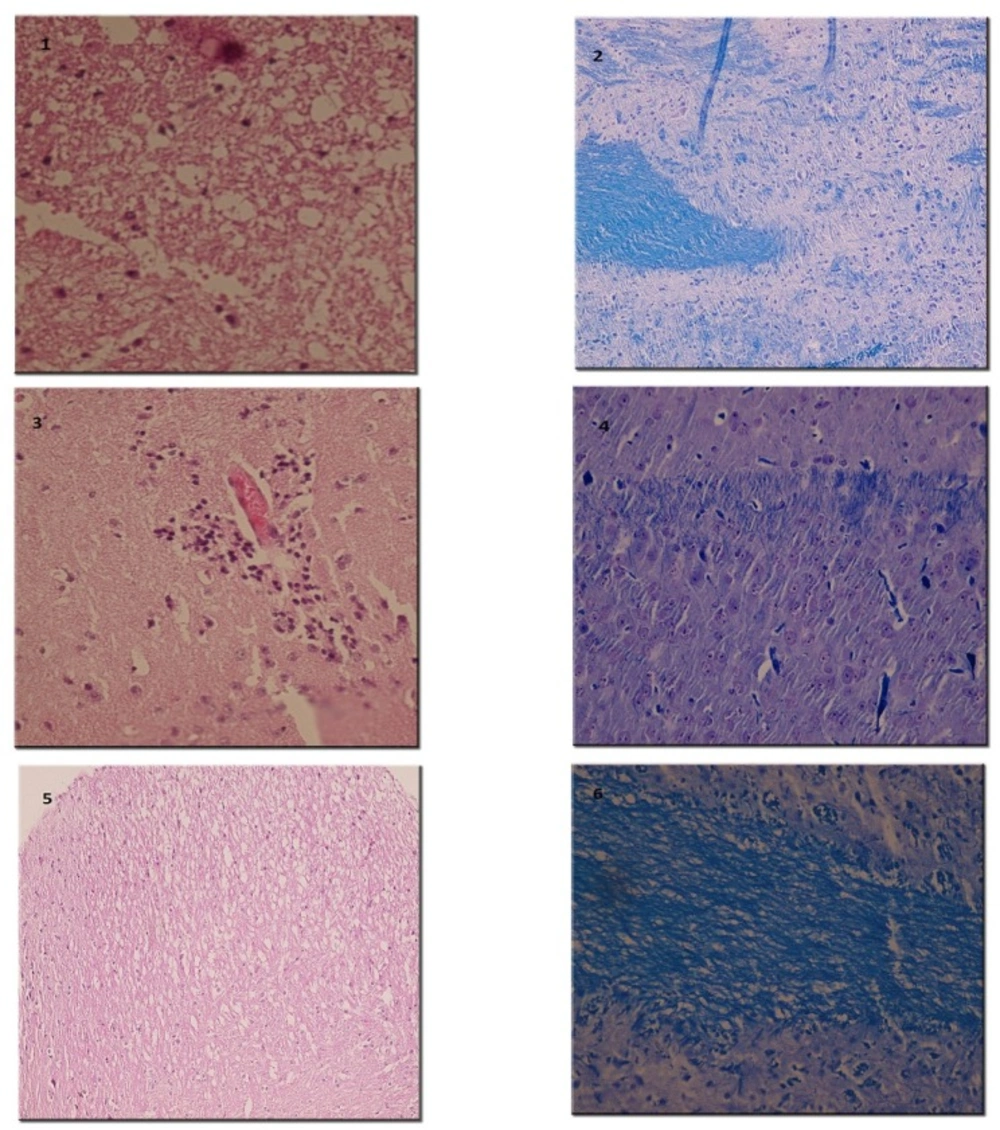
Pathological study of spinal cords from distinct groups revealed different extents of inflammation, myeline degradation and axoinal loss. Inflammation was significantly lower in the spinal cords of the RO 25-6981-treated EAE-mice. High-dose RO 25-6981 effectively resolved infiltration of inflammatory cells in the spinal cords of the EAE mice. Memantine was less effective in decreasing inflammation. This suggests superior efficacy of NR2B inhibition compared to non-specific antagonisation of NMDARs. RO 25-6981 was dose dependently effective in decreasing myelin degeneration. High dose of RO 25-6981 decreased demyelination significantly more than low dose. Compared with memantine, however, this therapeutic effect was not significantly different.
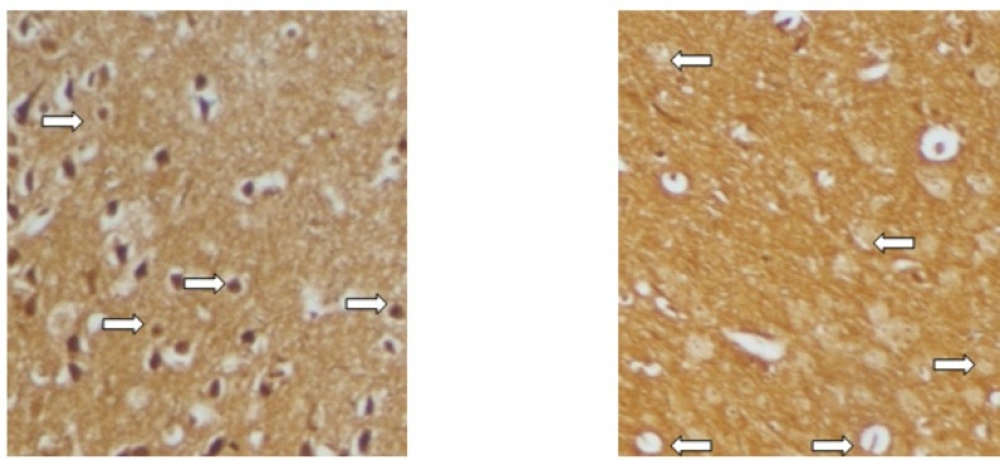
Sevierity of axonal degeneration was less in the groups treated with RO 25-6981. This highly selective antagonist shows efficacy in decreasing neuroaxonal injury or prevention of axonal loss. This effect can be mechanistically attributed to modulation of excitotoxicity. NR2B-containing NMDARs are believed to play a pivotal role in excitotoxicity (
43). The therapeutic effect of RO 25-6981 can be secondary to modulation of NR2B-containing NMDARs. The stimulatory effect of RO 25-6981 on neurogenesis could also be a reason (
44).
Moreover, the effect of RO 25-6981 could be partially attributable to decrease of inflammation in the CNS. This inflammation-decreasing effect could be a consequence of modulation of BBB disruption by NR2B-containing NMDAR inhibition. Excitotoxicity has been proposed to contribute to the pathogenesis of BBB break-down via NMDARs in endothelial cells (
7,
45). Our study supports this idea, because modulation of EAE course and axonal loss by the specific antagonist of NR2B subtype is simultaneous with dramatic resolution of inflammatory cell infiltration in the spinal cord. There are no published reports studying the effect of NR2B-containing NMDARs on BBB disruption in EAE, and the validity of this probable mechanism needs to be investigated independently.
Compared to memantine, RO 25-6981 appears to be more protective against EAE disease progression and histopathological evidence regarding inflammation and axonal degeneration. The superior efficacy of Ro 25-6981 is attributable to the pharmacological characteristics of this selective antagonist: much higher efficiency of RO 25-6981 in blocking NR2B-containing NMDARs, lack of inhibitory effect on NR2A-containing NMDARs, and its higher potency compared to memantine (
23,
46).
It is reported here that short-term treatment of EAE with antagonists of NR2B subtype could be effective in terms of disease modulation and pathological changes. Long-term effects remain to be studied. The biological biomarkers of axonal loss and cell death can be followed in other studies for further evaluation of the mechanism of action. Preventive administration of Ro 25-6981 can also help in understanding the role of NR2B-containing NMDARs in EAE development, as a prerequisite to proposing hypotheses about human MS. Our findings could be used as a cornerstone for hypothesizing theories and designing complementary studies to validate NR2B-containing NMDARs as possible targets in pharmacotherapy of MS.